

Genomic and Single-Cell Landscape Reveals Novel Drivers and Therapeutic Vulnerabilities of Transformed Cutaneous T-cell Lymphoma
- PMID: 35247891
- PMCID: PMC9148441
- DOI: 10.1158/2159-8290.CD-21-1207
Genomic and Single-Cell Landscape Reveals Novel Drivers and Therapeutic Vulnerabilities of Transformed Cutaneous T-cell Lymphoma
Abstract
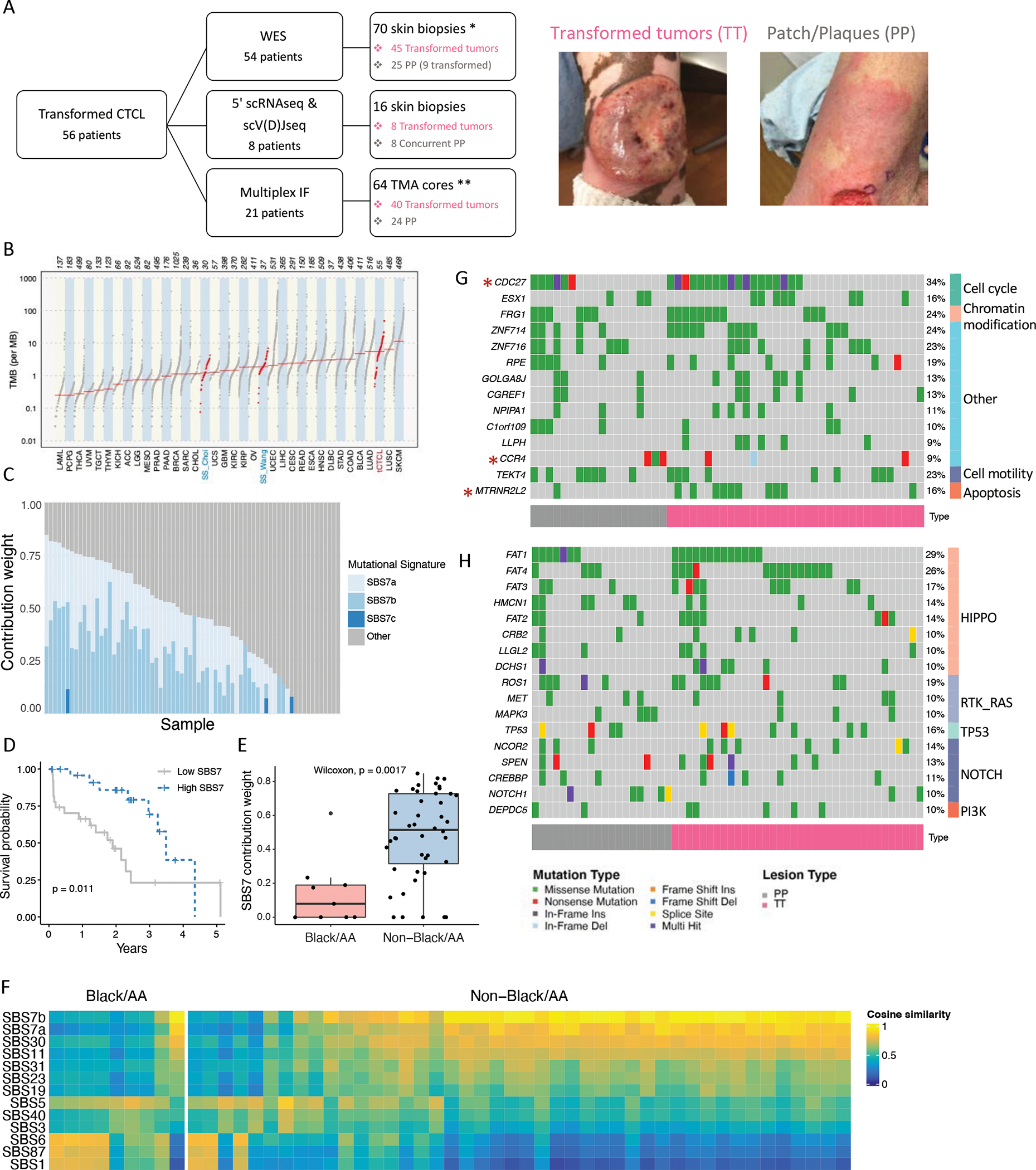
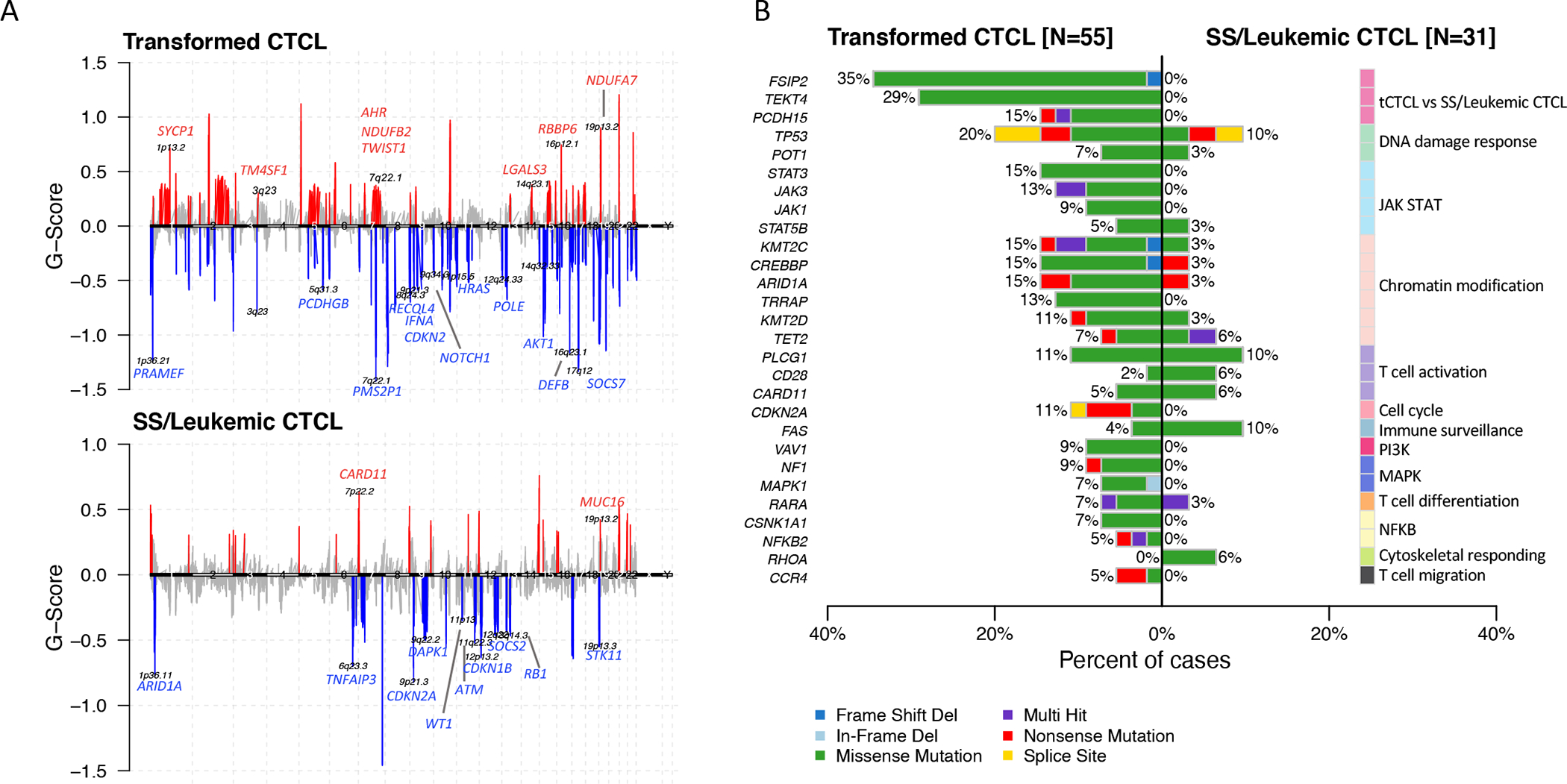
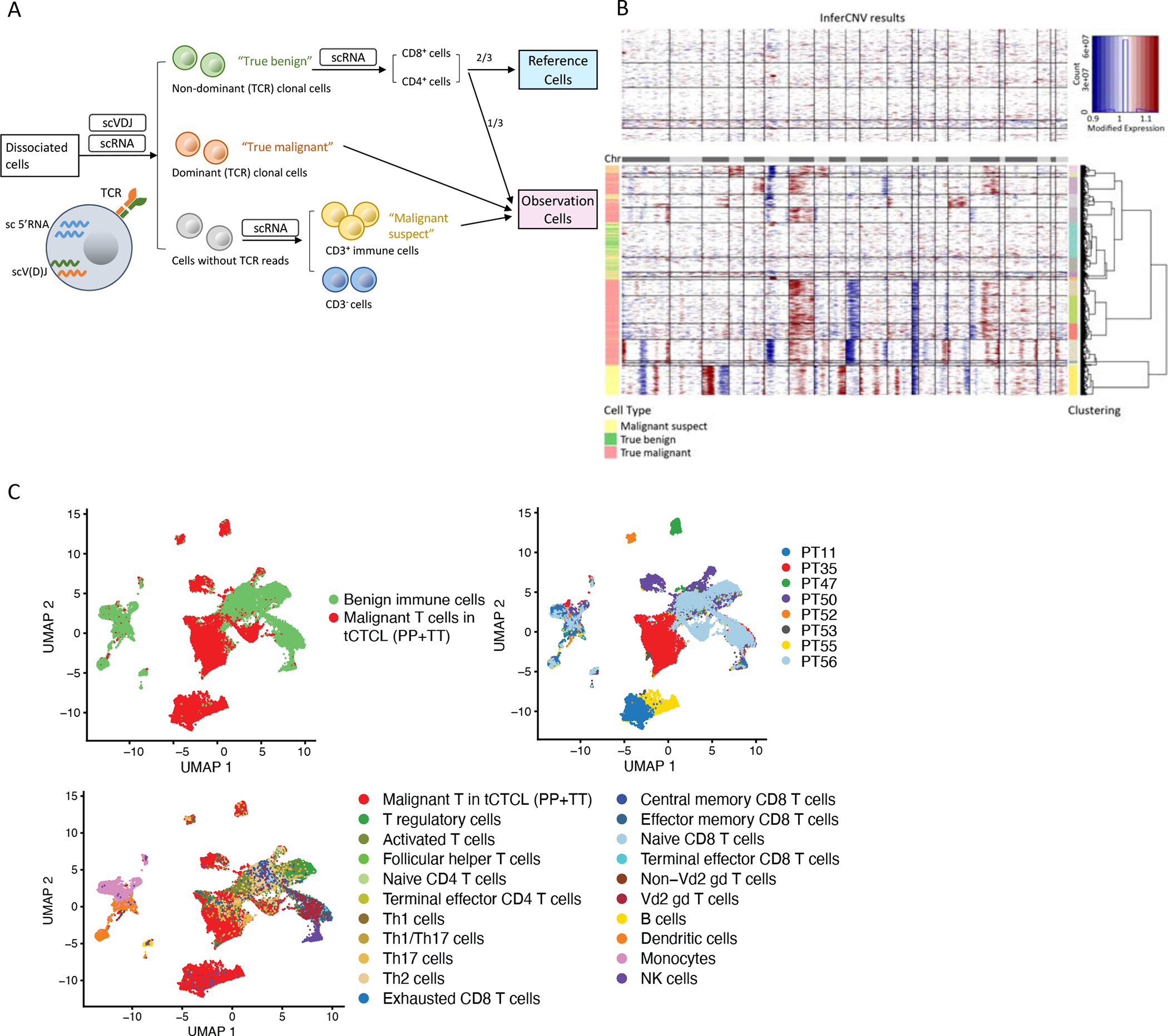
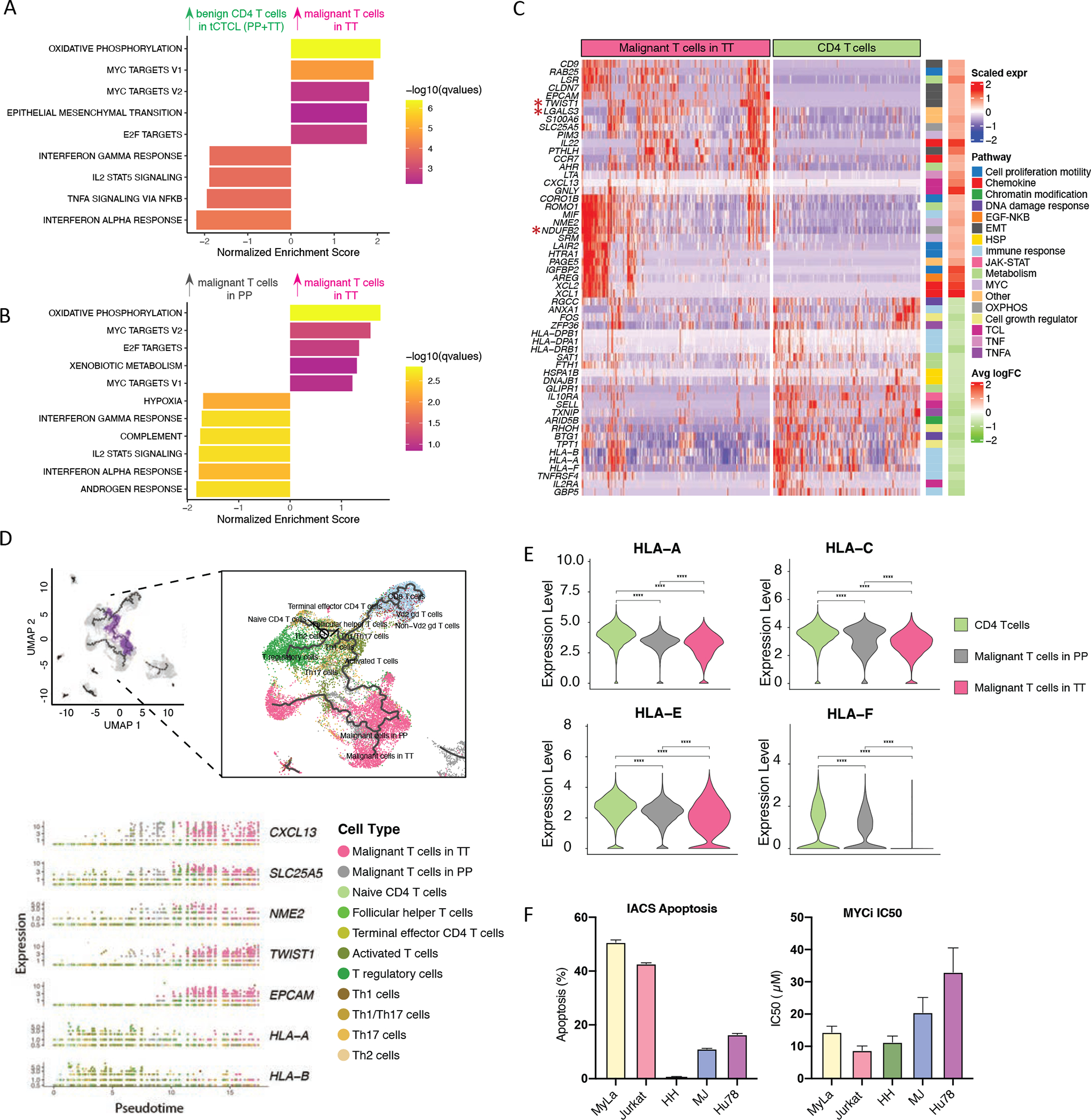
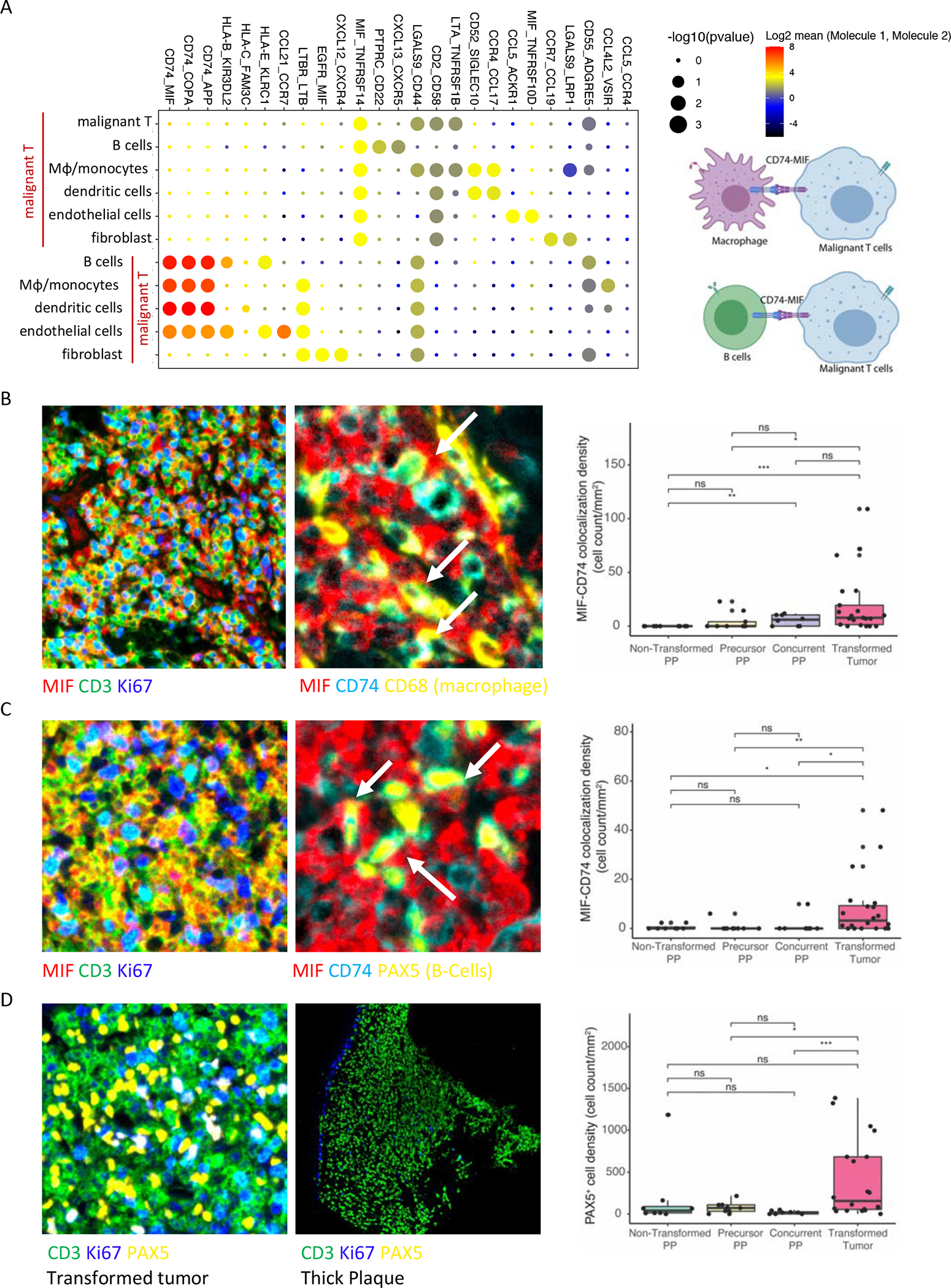
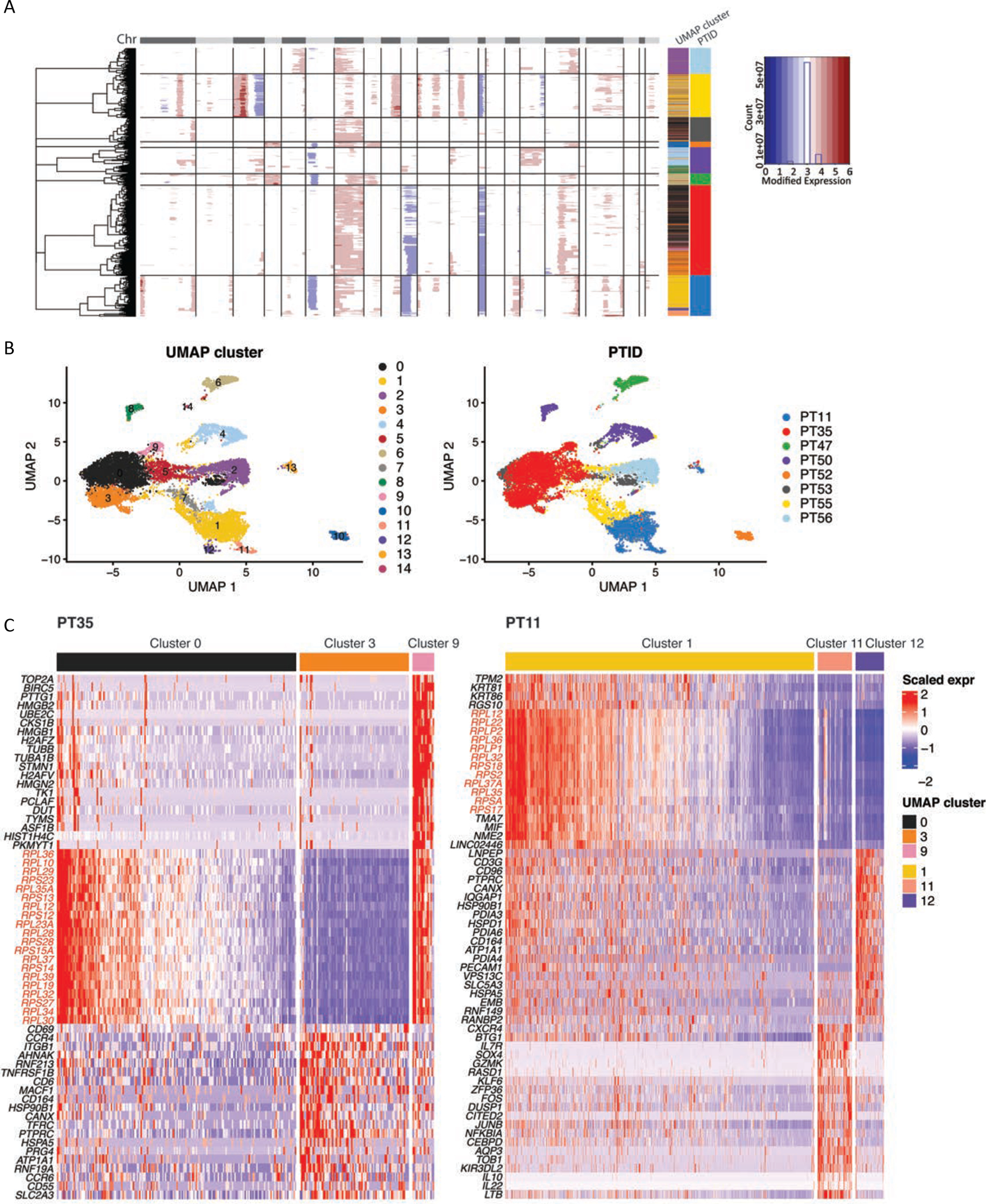
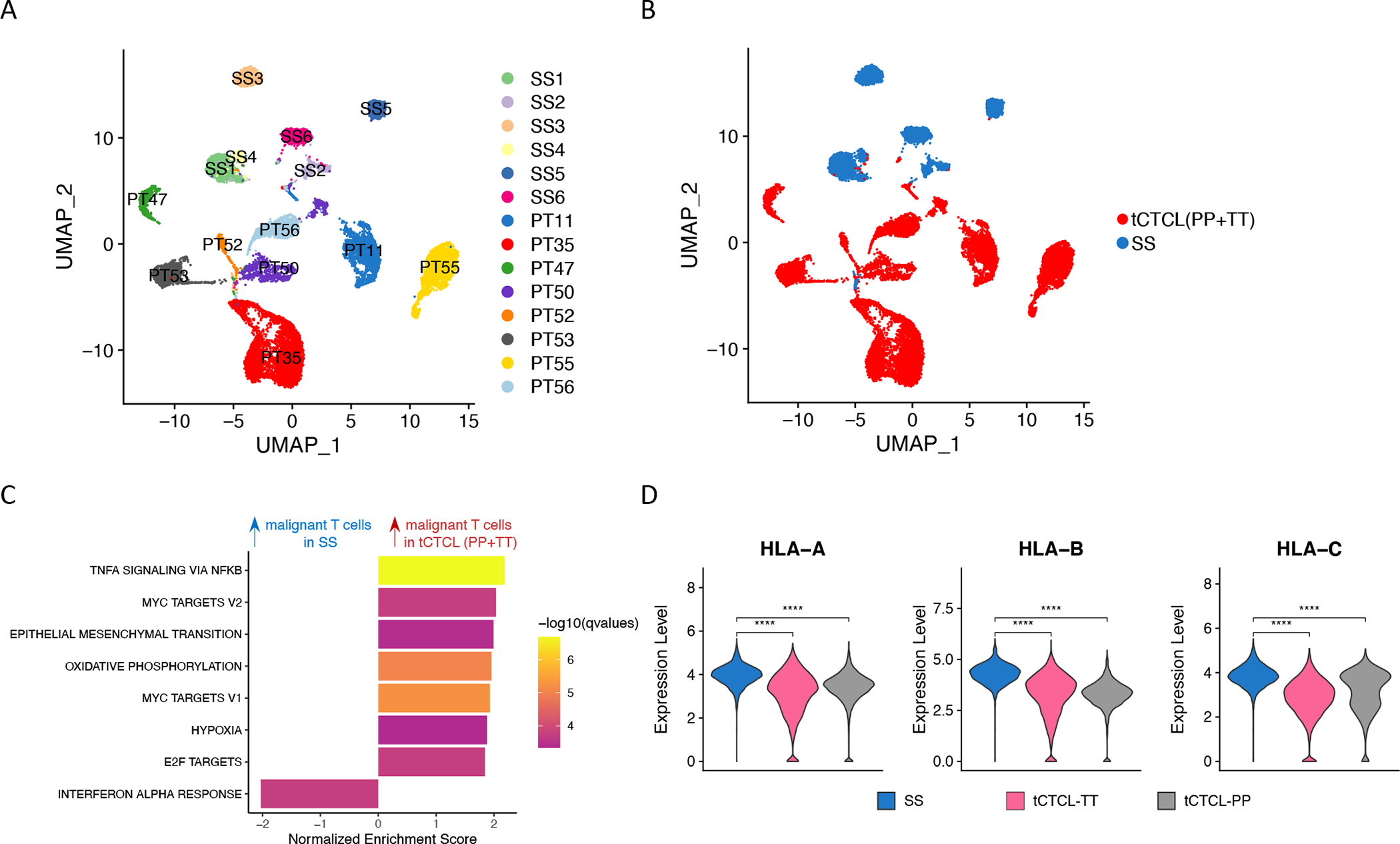
Abstract: Cutaneous T-cell lymphoma (CTCL) is a rare cancer of skin-homing T cells. A subgroup of patients develops large cell transformation with rapid progression to an aggressive lymphoma. Here, we investigated the transformed CTCL (tCTCL) tumor ecosystem using integrative multiomics spanning whole-exome sequencing (WES), single-cell RNA sequencing, and immune profiling in a unique cohort of 56 patients. WES of 70 skin biopsies showed high tumor mutation burden, UV signatures that are prognostic for survival, exome-based driver events, and most recurrently mutated pathways in tCTCL. Single-cell profiling of 16 tCTCL skin biopsies identified a core oncogenic program with metabolic reprogramming toward oxidative phosphorylation (OXPHOS), cellular plasticity, upregulation of MYC and E2F activities, and downregulation of MHC I suggestive of immune escape. Pharmacologic perturbation using OXPHOS and MYC inhibitors demonstrated potent antitumor activities, whereas immune profiling provided in situ evidence of intercellular communications between malignant T cells expressing macrophage migration inhibitory factor and macrophages and B cells expressing CD74.
Significance: Our study contributes a key resource to the community with the largest collection of tCTCL biopsies that are difficult to obtain. The multiomics data herein provide the first comprehensive compendium of genomic alterations in tCTCL and identify potential prognostic signatures and novel therapeutic targets for an incurable T-cell lymphoma. This article is highlighted in the In This Issue feature, p. 1171.
©2022 American Association for Cancer Research.
Conflict of interest statement
Conflicts of interest disclosures
L.S.V. or her institution received research support from Helsinn, Eisai Co., Soligenix, Kyowa Kirin Inc., Innate Pharma, and Elorac Inc. for CTCL studies. L.S.V. is a consultant and speaker for Kyowa Kirin Inc. and Helsinn. L.S. is advisory board member of Kyowa Kirin and consultant at Dren-Bio. S.W. is advisory board member and KOL for Kyowa Kirin.
Figures
References
-
- Scarisbrick JJ, Prince HM, Vermeer MH, Quaglino P, Horwitz S, Porcu P, et al. Cutaneous Lymphoma International Consortium Study of Outcome in Advanced Stages of Mycosis Fungoides and Sezary Syndrome: Effect of Specific Prognostic Markers on Survival and Development of a Prognostic Model. J Clin Oncol 2015;33(32):3766–73 doi 10.1200/JCO.2015.61.7142. - DOI - PMC - PubMed
-
- Jawed SI, Myskowski PL, Horwitz S, Moskowitz A, Querfeld C. Primary cutaneous T-cell lymphoma (mycosis fungoides and Sezary syndrome): part I. Diagnosis: clinical and histopathologic features and new molecular and biologic markers. J Am Acad Dermatol 2014;70(2):205 e1–16; quiz 21–2 doi 10.1016/j.jaad.2013.07.049. - DOI - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
