

Concurrent Inhibition of ERK and Farnesyltransferase Suppresses the Growth of HRAS Mutant Head and Neck Squamous Cell Carcinoma
- PMID: 35247914
- PMCID: PMC9081222
- DOI: 10.1158/1535-7163.MCT-21-0142
Concurrent Inhibition of ERK and Farnesyltransferase Suppresses the Growth of HRAS Mutant Head and Neck Squamous Cell Carcinoma
Abstract
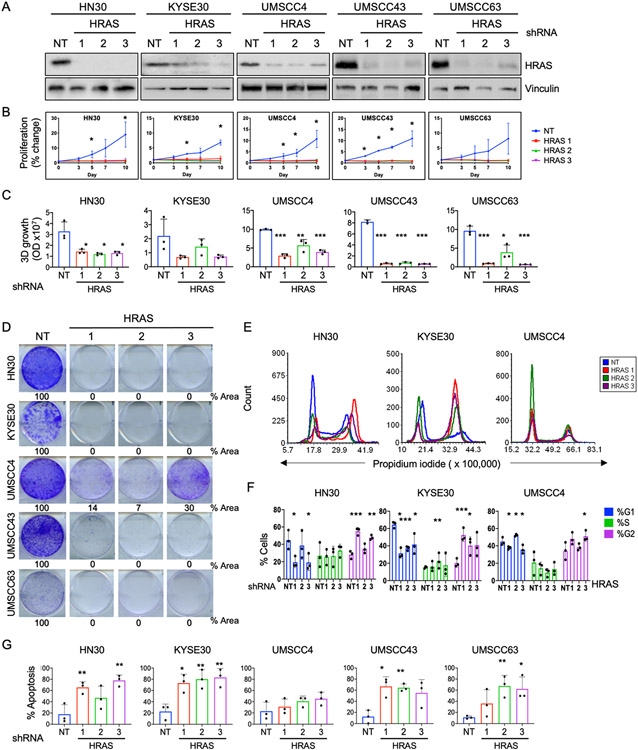
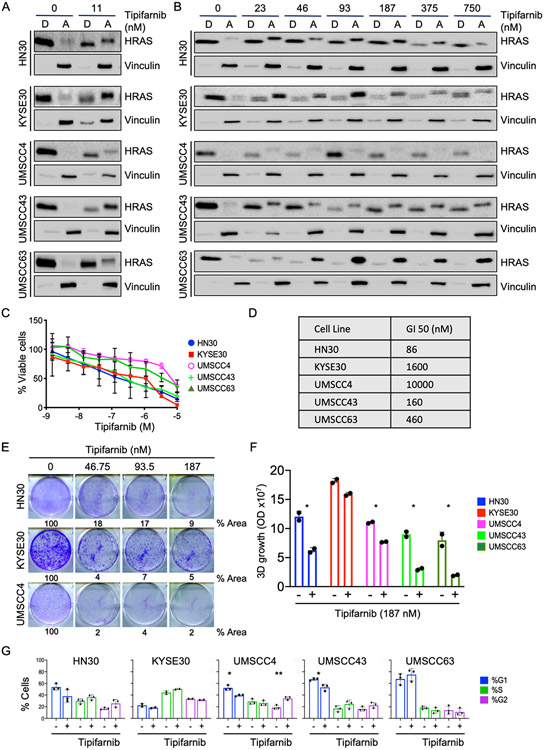
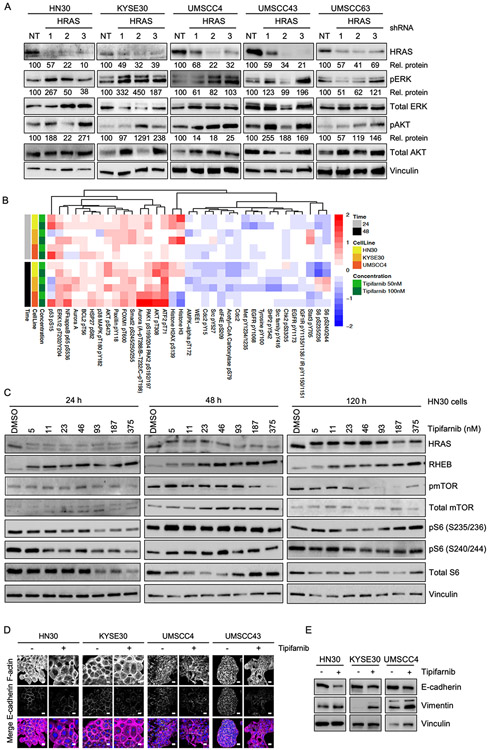
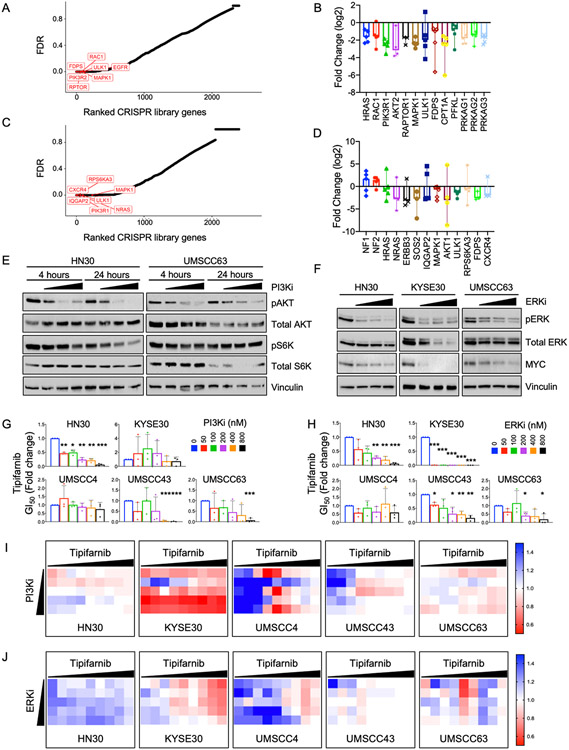
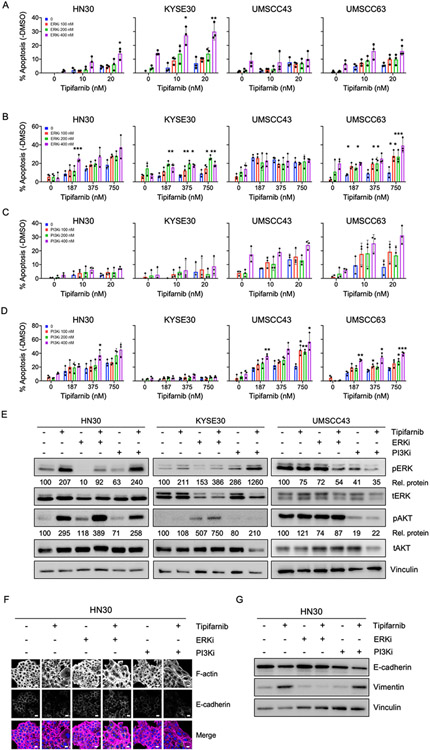
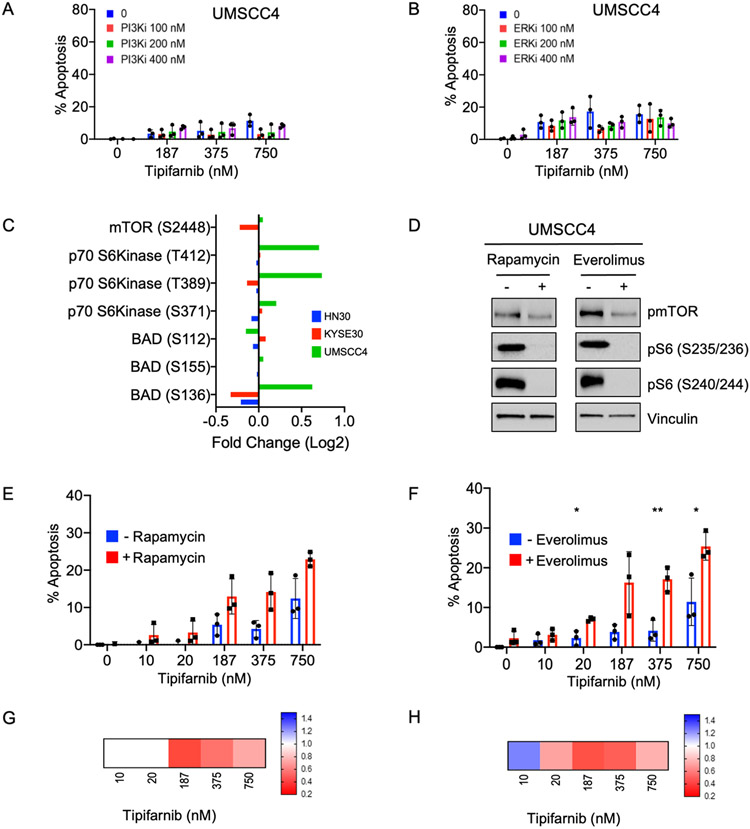
Human papilloma virus (HPV)-negative head and neck squamous cell carcinoma (HNSCC) is a common cancer worldwide with an unmet need for more effective, less toxic treatments. Currently, both the disease and the treatment of HNSCC cause significant mortality and morbidity. Targeted therapies hold new promise for patients with HPV-negative status whose tumors harbor oncogenic HRAS mutations. Recent promising clinical results have renewed interest in the development of farnesyltransferase inhibitors (FTIs) as a therapeutic strategy for HRAS-mutant cancers. With the advent of clinical evaluation of the FTI tipifarnib for the treatment of HRAS-mutant HNSCC, we investigated the activity of tipifarnib and inhibitors of HRAS effector signaling in HRAS-mutant HNSCC cell lines. First, we validated that HRAS is a cancer driver in HRAS-mutant HNSCC lines. Second, we showed that treatment with the FTI tipifarnib largely phenocopied HRAS silencing, supporting HRAS as a key target of FTI antitumor activity. Third, we performed reverse-phase protein array analyses to profile FTI treatment-induced changes in global signaling, and conducted CRISPR/Cas9 genetic loss-of-function screens to identify previously unreported genes and pathways that modulate sensitivity to tipifarnib. Fourth, we determined that concurrent inhibition of HRAS effector signaling (ERK, PI3K, mTORC1) increased sensitivity to tipifarnib treatment, in part by overcoming tipifarnib-induced compensatory signaling. We also determined that ERK inhibition could block tipifarnib-induced epithelial-to-mesenchymal transition, providing a potential basis for the effectiveness of this combination. Our results support future investigations of these and other combination treatments for HRAS mutant HNSCC.
©2022 American Association for Cancer Research.
Conflict of interest statement
Conflict of interest statement:
M. Pierobon and EF Petricoin III are inventors on US Government and University assigned patents and patent applications. As inventors, they are entitled to receive royalties as provided by US Law and George Mason University policy. They receive royalties from TheraLink Technologies and are consultants to and/or shareholders of Avant Diagnostics, Inc. EF Petricoin III is also consultant and shareholder of Perthera, Inc. CJ Der is a consultant/advisory board member for Mirati Therapeutics, Deciphera 950 Pharmaceuticals and Anchiano Therapeutics. CJ Der has received research funding support from Mirati Therapeutics and Deciphera Pharmaceuticals. CJ Der has consulted for Ribometrix, Jazz Therapeutics, SVB Leerink, Axon Advisors LLC, Third Bridge, Sanofi, SmartAnalyst, Turning Point Therapeutics and Eli Lilly. AD Cox has consulted for Eli Lilly and Mirati Therapeutics. The remaining authors declare no conflicts of interest.
Figures
References
-
- End DW, Smets G, Todd AV, Applegate TL, Fuery CJ, Angibaud P, et al. Characterization of the antitumor effects of the selective farnesyl protein transferase inhibitor R115777 in vivo and in vitro. Cancer Res 2001;61:131–7 - PubMed
-
- Liu M, Bryant MS, Chen J, Lee S, Yaremko B, Lipari P, et al. Antitumor activity of SCH 66336, an orally bioavailable tricyclic inhibitor of farnesyl protein transferase, in human tumor xenograft models and wap-ras transgenic mice. Cancer Res 1998;58:4947–56 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
