

Incorporating Within-Host Diversity in Phylogenetic Analyses for Detecting Clusters of New HIV Diagnoses
- PMID: 35250908
- PMCID: PMC8891961
- DOI: 10.3389/fmicb.2021.803190
Incorporating Within-Host Diversity in Phylogenetic Analyses for Detecting Clusters of New HIV Diagnoses
Abstract
Background: Phylogenetic analyses of HIV sequences are used to detect clusters and inform public health interventions. Conventional approaches summarize within-host HIV diversity with a single consensus sequence per host of the pol gene, obtained from Sanger or next-generation sequencing (NGS). There is growing recognition that this approach discards potentially important information about within-host sequence variation, which can impact phylogenetic inference. However, whether alternative summary methods that incorporate intra-host variation impact phylogenetic inference of transmission network features is unknown.
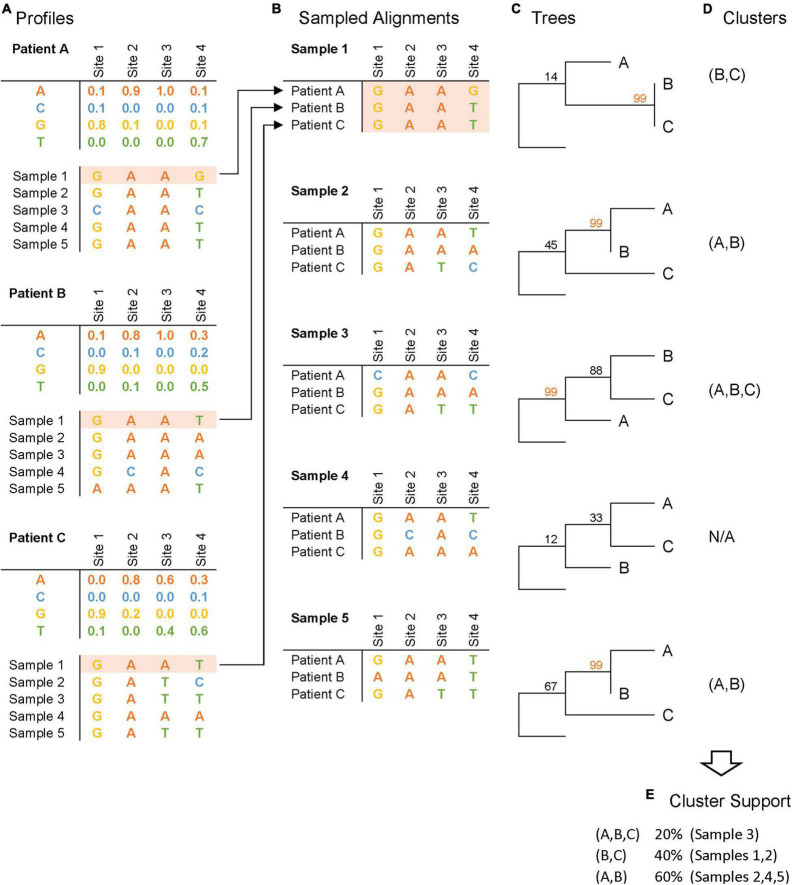
Methods: We introduce profile sampling, a method to incorporate within-host NGS sequence diversity into phylogenetic HIV cluster inference. We compare this approach to Sanger- and NGS-derived pol and near-whole-genome consensus sequences and evaluate its potential benefits in identifying molecular clusters among all newly-HIV-diagnosed individuals over six months at the largest HIV center in Rhode Island.
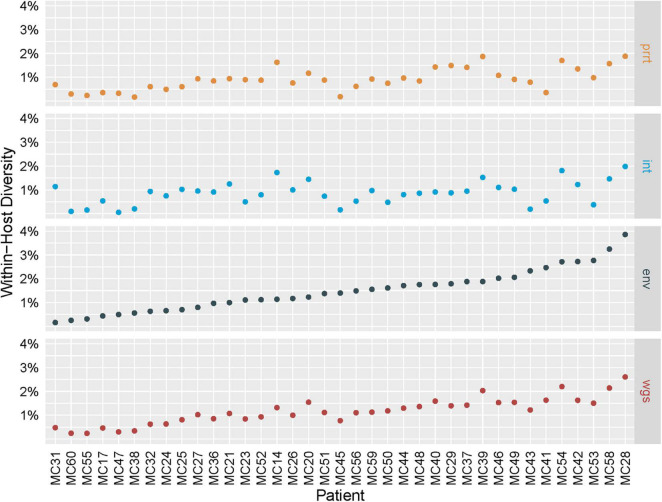
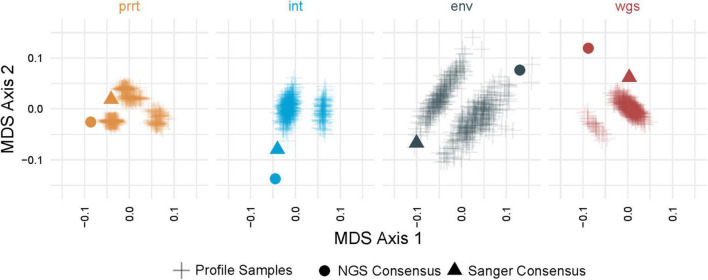
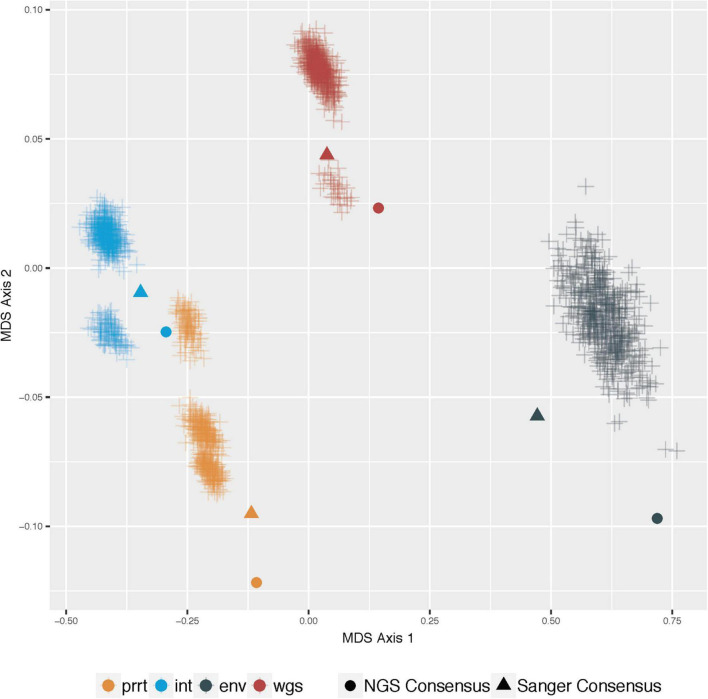
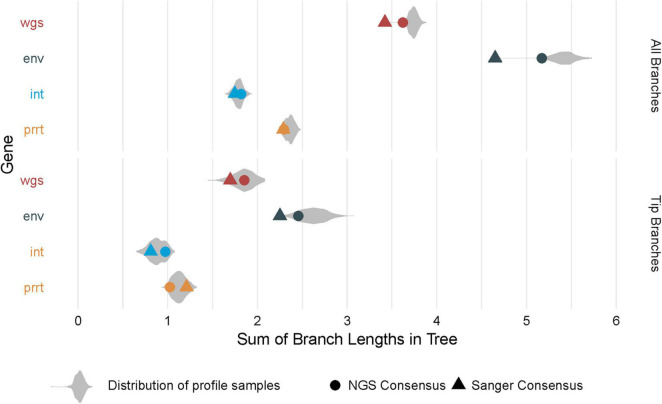
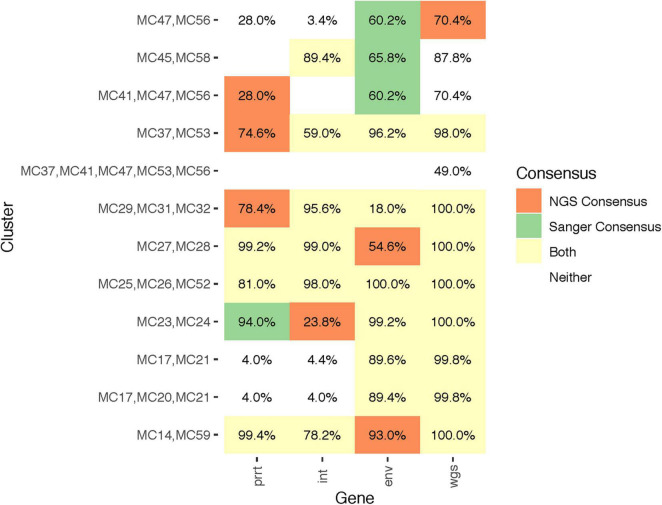
Results: Profile sampling cluster inference demonstrated that within-host viral diversity impacts phylogenetic inference across individuals, and that consensus sequence approaches can obscure both magnitude and effect of these impacts. Clustering differed between Sanger- and NGS-derived consensus and profile sampling sequences, and across gene regions.
Discussion: Profile sampling can incorporate within-host HIV diversity captured by NGS into phylogenetic analyses. This additional information can improve robustness of cluster detection.
Keywords: HIV; cluster inference; consensus sequence; near-whole-genome; next generation sequencing (NGS); phylogenetics; profile sampling; transmission disruption.
Copyright © 2022 Guang, Howison, Ledingham, D’Antuono, Chan, Lawrence, Dunn and Kantor.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures


References
-
- Billera L. J., Holmes S. P., Vogtmann K. (2001). Geometry of the space of phylogenetic trees. Adv. Appl. Math. 27 733–767.
-
- Eddy S. R. (2004). What is a hidden Markov model? Nat. Biotechnol. 22 1315–1316. - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources