

In Vivo Targeting Replication Protein A for Cancer Therapy
- PMID: 35251993
- PMCID: PMC8895377
- DOI: 10.3389/fonc.2022.826655
In Vivo Targeting Replication Protein A for Cancer Therapy
Abstract
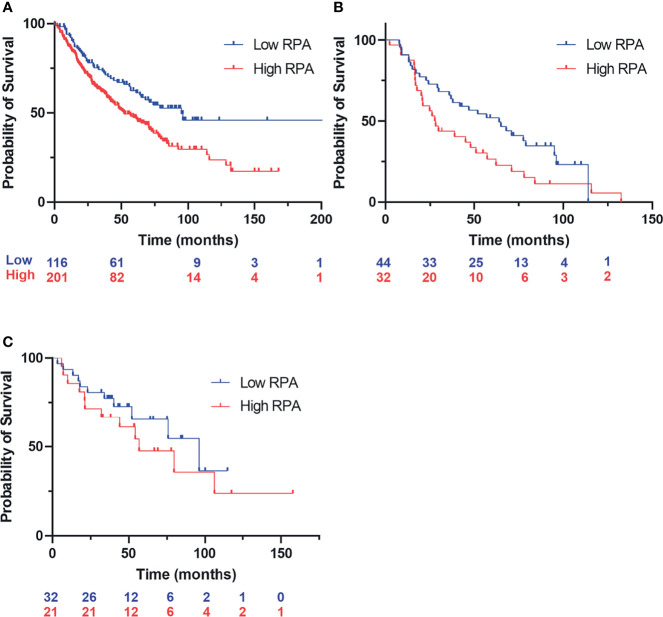
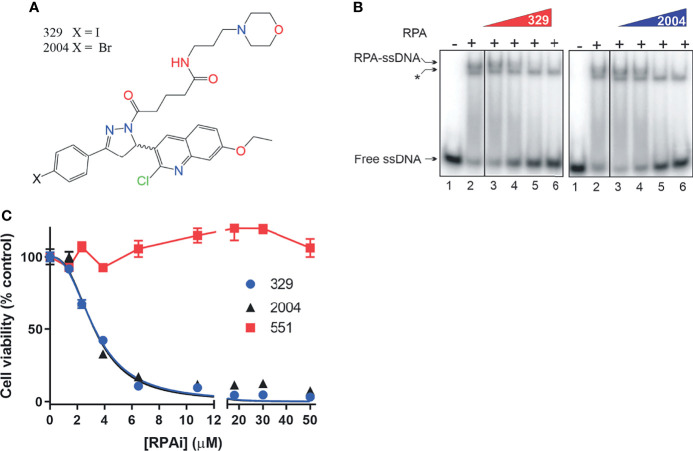
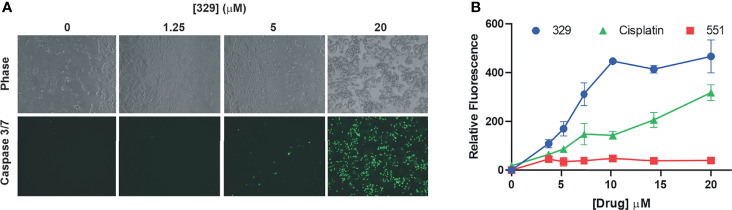
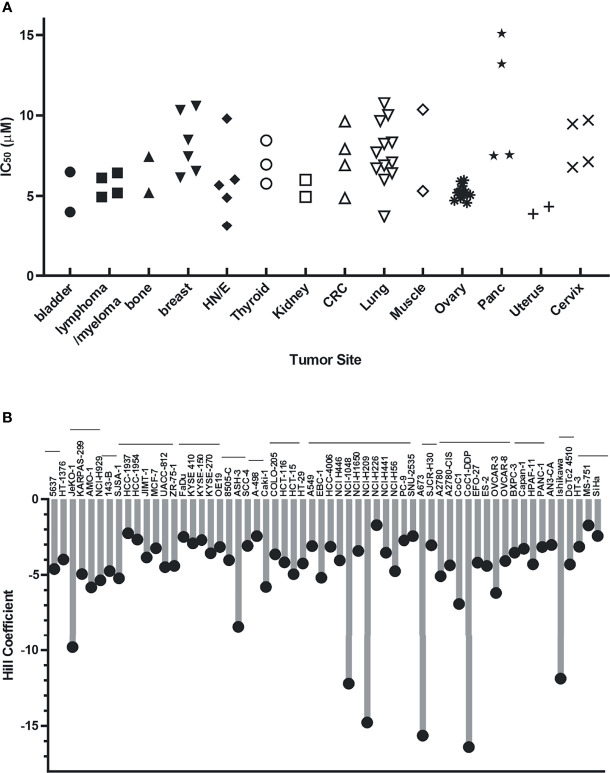
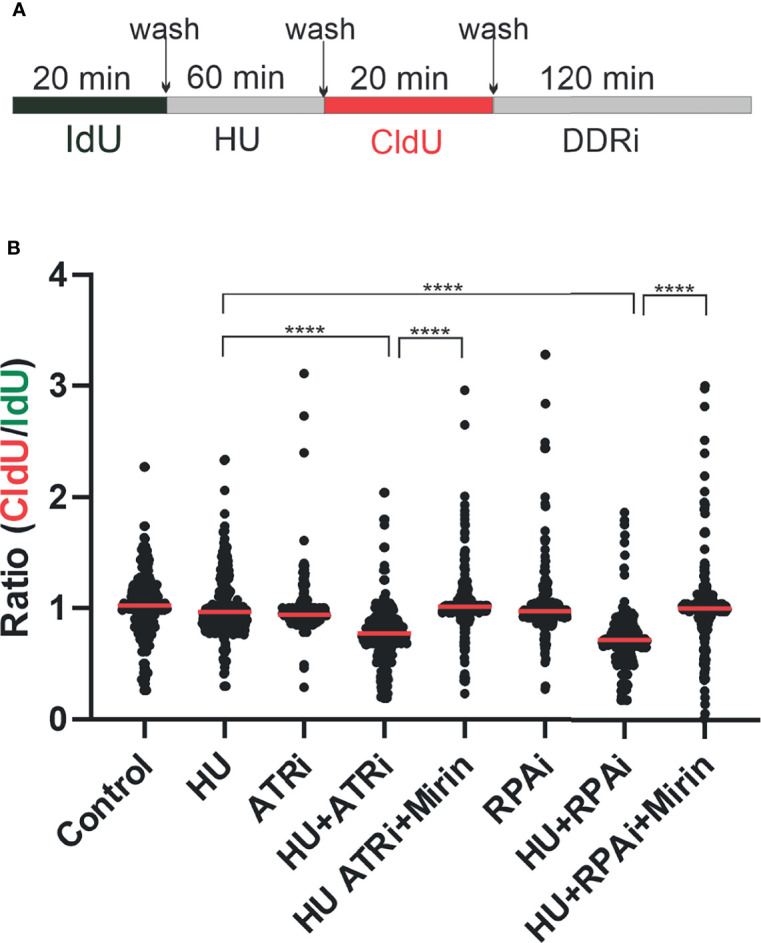
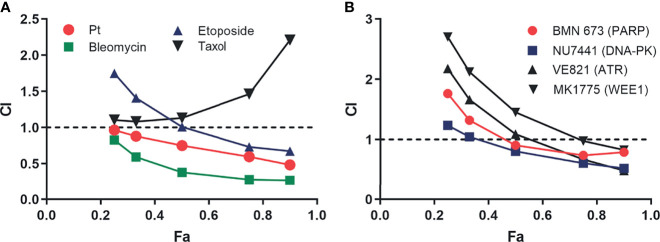
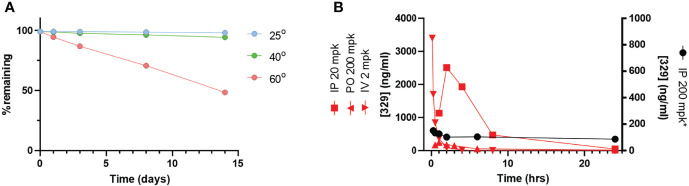
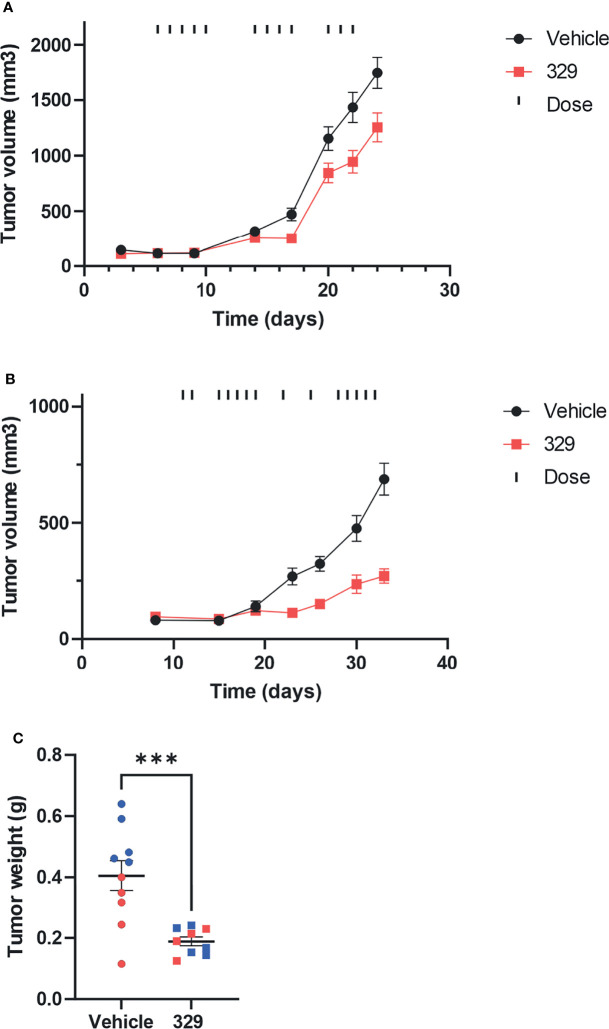
Replication protein A (RPA) plays essential roles in DNA replication, repair, recombination, and the DNA damage response (DDR). Retrospective analysis of lung cancer patient data demonstrates high RPA expression as a negative prognostic biomarker for overall survival in smoking-related lung cancers. Similarly, relative expression of RPA is a predictive marker for response to chemotherapy. These observations are consistent with the increase in RPA expression serving as an adaptive mechanism that allows tolerance of the genotoxic stress resulting from carcinogen exposure. We have developed second-generation RPA inhibitors (RPAis) that block the RPA-DNA interaction and optimized formulation for in vivo analyses. Data demonstrate that unlike first-generation RPAis, second-generation molecules show increased cellular permeability and induce cell death via apoptosis. Second-generation RPAis elicit single-agent in vitro anticancer activity across a broad spectrum of cancers, and the cellular response suggests existence of a threshold before chemical RPA exhaustion induces cell death. Chemical RPA inhibition potentiates the anticancer activity of a series of DDR inhibitors and traditional DNA-damaging cancer therapeutics. Consistent with chemical RPA exhaustion, we demonstrate that the effects of RPAi on replication fork dynamics are similar to other known DDR inhibitors. An optimized formulation of RPAi NERx 329 was developed that resulted in single-agent anticancer activity in two non-small cell lung cancer models. These data demonstrate a unique mechanism of action of RPAis eliciting a state of chemical RPA exhaustion and suggest they will provide an effective therapeutic option for difficult-to-treat lung cancers.
Keywords: DNA damage response; DNA repair and cancer; DNA repair inhibitors; Replication Protein A; Replication Stress Response.
Copyright © 2022 VanderVere-Carozza, Gavande, Jalal, Pollok, Ekinci, Heyza, Patrick, Masters, Turchi and Pawelczak.
Conflict of interest statement
Author JT is a shareholder and founder and KP is a shareholder and employed by NERx BioSciences. The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
References
Grants and funding
LinkOut - more resources
Full Text Sources