

STRIDE: accurately decomposing and integrating spatial transcriptomics using single-cell RNA sequencing
- PMID: 35253896
- PMCID: PMC9023289
- DOI: 10.1093/nar/gkac150
STRIDE: accurately decomposing and integrating spatial transcriptomics using single-cell RNA sequencing
Erratum in
-
Correction to 'STRIDE: accurately decomposing and integrating spatial transcriptomics using single-cell RNA sequencing'.Nucleic Acids Res. 2022 Apr 22;50(7):4198. doi: 10.1093/nar/gkac254. Nucleic Acids Res. 2022. PMID: 35390158 Free PMC article. No abstract available.
Abstract
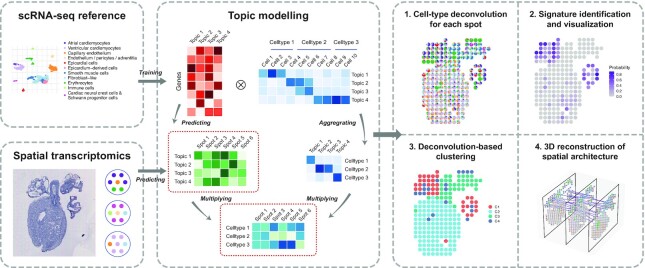
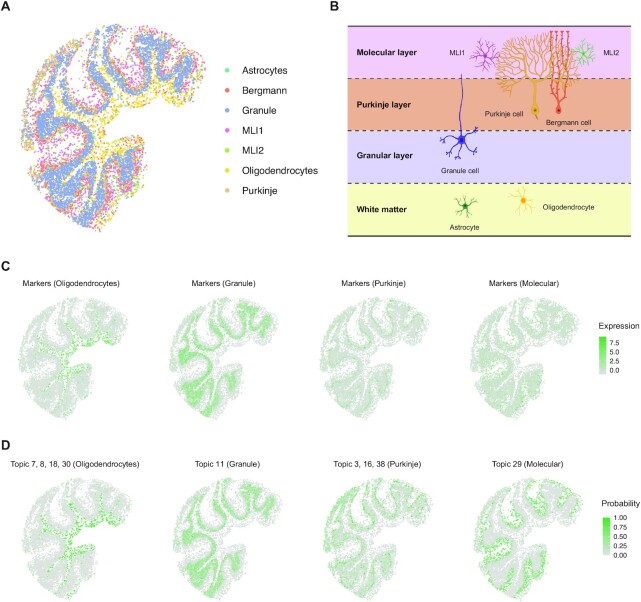
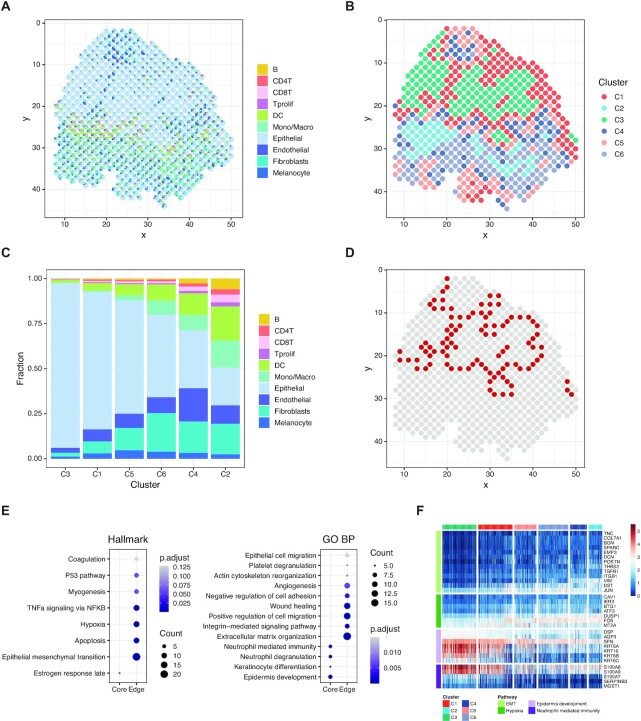
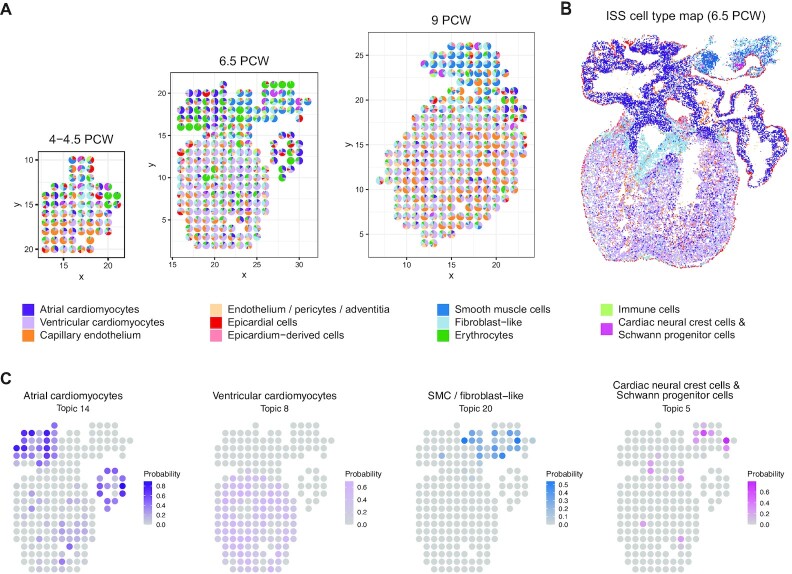
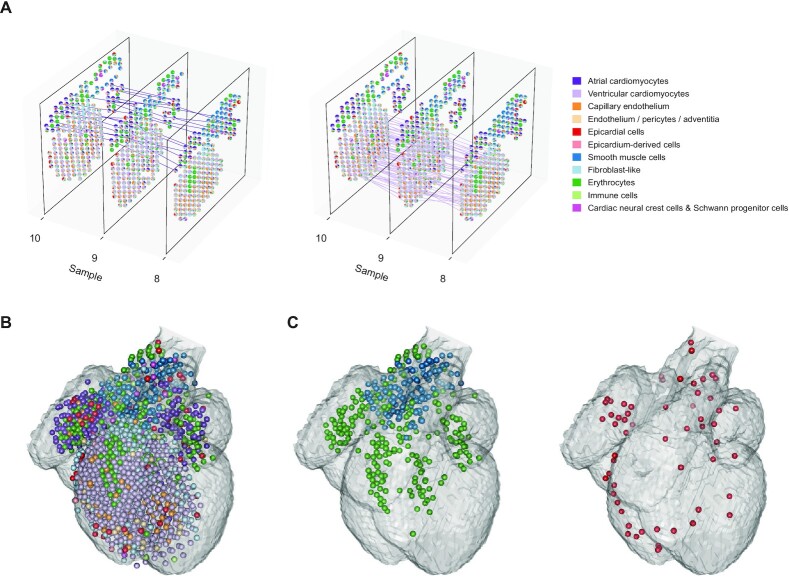
The recent advances in spatial transcriptomics have brought unprecedented opportunities to understand the cellular heterogeneity in the spatial context. However, the current limitations of spatial technologies hamper the exploration of cellular localizations and interactions at single-cell level. Here, we present spatial transcriptomics deconvolution by topic modeling (STRIDE), a computational method to decompose cell types from spatial mixtures by leveraging topic profiles trained from single-cell transcriptomics. STRIDE accurately estimated the cell-type proportions and showed balanced specificity and sensitivity compared to existing methods. We demonstrated STRIDE's utility by applying it to different spatial platforms and biological systems. Deconvolution by STRIDE not only mapped rare cell types to spatial locations but also improved the identification of spatially localized genes and domains. Moreover, topics discovered by STRIDE were associated with cell-type-specific functions and could be further used to integrate successive sections and reconstruct the three-dimensional architecture of tissues. Taken together, STRIDE is a versatile and extensible tool for integrated analysis of spatial and single-cell transcriptomics and is publicly available at https://github.com/wanglabtongji/STRIDE.
© The Author(s) 2022. Published by Oxford University Press on behalf of Nucleic Acids Research.
Figures

Similar articles
-
Robust decomposition of cell type mixtures in spatial transcriptomics.Nat Biotechnol. 2022 Apr;40(4):517-526. doi: 10.1038/s41587-021-00830-w. Epub 2021 Feb 18. Nat Biotechnol. 2022. PMID: 33603203 Free PMC article.
-
LETSmix: a spatially informed and learning-based domain adaptation method for cell-type deconvolution in spatial transcriptomics.Genome Med. 2025 Feb 28;17(1):16. doi: 10.1186/s13073-025-01442-8. Genome Med. 2025. PMID: 40022231 Free PMC article.
-
De novo analysis of bulk RNA-seq data at spatially resolved single-cell resolution.Nat Commun. 2022 Oct 30;13(1):6498. doi: 10.1038/s41467-022-34271-z. Nat Commun. 2022. PMID: 36310179 Free PMC article.
-
Encoding Method of Single-cell Spatial Transcriptomics Sequencing.Int J Biol Sci. 2020 Jul 30;16(14):2663-2674. doi: 10.7150/ijbs.43887. eCollection 2020. Int J Biol Sci. 2020. PMID: 32792863 Free PMC article. Review.
-
Integrating single-cell and spatial transcriptomics to elucidate intercellular tissue dynamics.Nat Rev Genet. 2021 Oct;22(10):627-644. doi: 10.1038/s41576-021-00370-8. Epub 2021 Jun 18. Nat Rev Genet. 2021. PMID: 34145435 Free PMC article. Review.
Cited by
-
Computational Strategies and Algorithms for Inferring Cellular Composition of Spatial Transcriptomics Data.Genomics Proteomics Bioinformatics. 2024 Sep 13;22(3):qzae057. doi: 10.1093/gpbjnl/qzae057. Genomics Proteomics Bioinformatics. 2024. PMID: 39110523 Free PMC article. Review.
-
Advances in spatial transcriptomics and its applications in cancer research.Mol Cancer. 2024 Jun 20;23(1):129. doi: 10.1186/s12943-024-02040-9. Mol Cancer. 2024. PMID: 38902727 Free PMC article. Review.
-
Accurate and efficient integrative reference-informed spatial domain detection for spatial transcriptomics.Nat Methods. 2024 Jul;21(7):1231-1244. doi: 10.1038/s41592-024-02284-9. Epub 2024 Jun 6. Nat Methods. 2024. PMID: 38844627 Free PMC article.
-
Deconvolution and inference of spatial communication through optimization algorithm for spatial transcriptomics.Commun Biol. 2025 Feb 14;8(1):235. doi: 10.1038/s42003-025-07625-8. Commun Biol. 2025. PMID: 39948133 Free PMC article.
-
Implementing distinct spatial proteogenomic technologies: opportunities, challenges, and key considerations.Clin Exp Immunol. 2024 Oct 16;218(2):151-162. doi: 10.1093/cei/uxae077. Clin Exp Immunol. 2024. PMID: 39133142 Free PMC article. Review.
References
-
- Junttila M.R., de Sauvage F.J.. Influence of tumour micro-environment heterogeneity on therapeutic response. Nature. 2013; 501:346–354. - PubMed
MeSH terms
LinkOut - more resources
Full Text Sources
