Accurate positioning of functional residues with robotics-inspired computational protein design
- PMID: 35254891
- PMCID: PMC8931229
- DOI: 10.1073/pnas.2115480119
Accurate positioning of functional residues with robotics-inspired computational protein design
Abstract
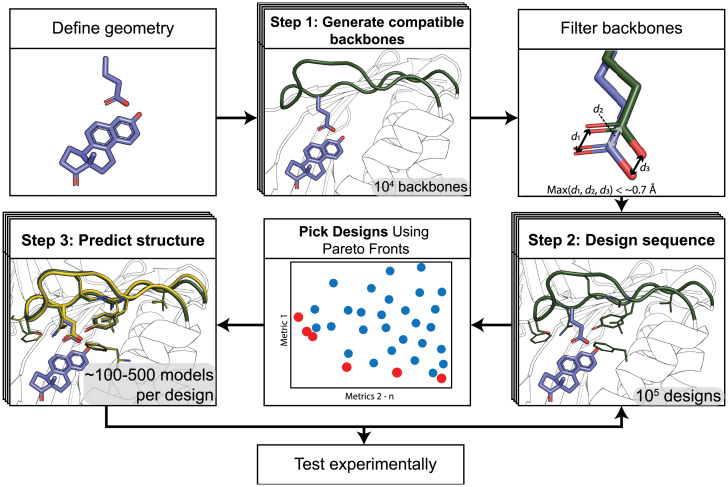
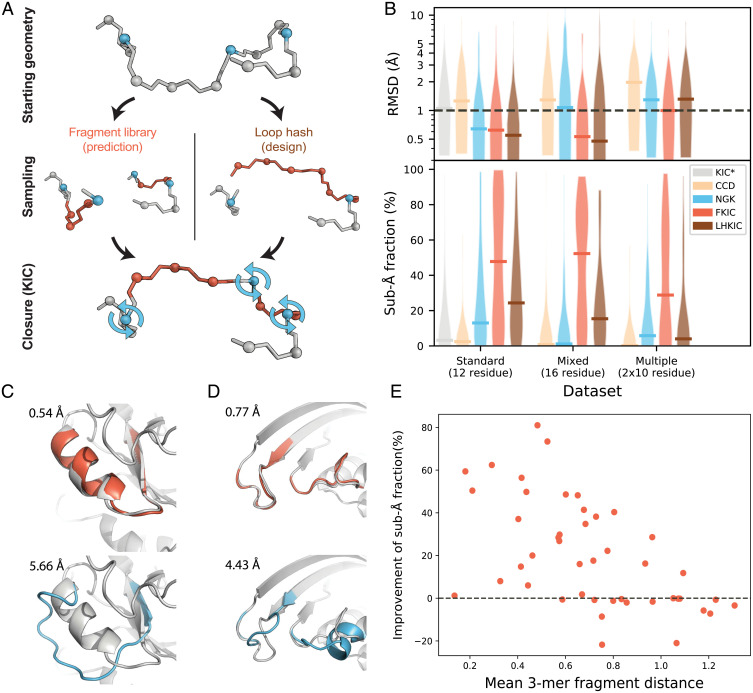
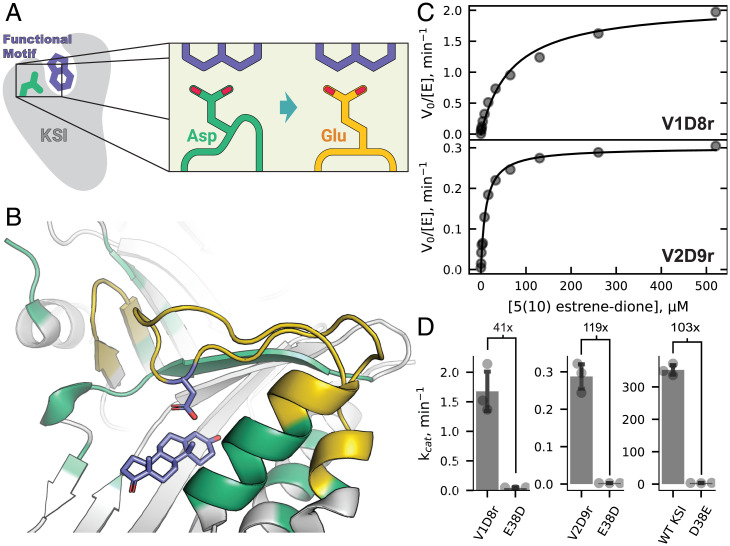
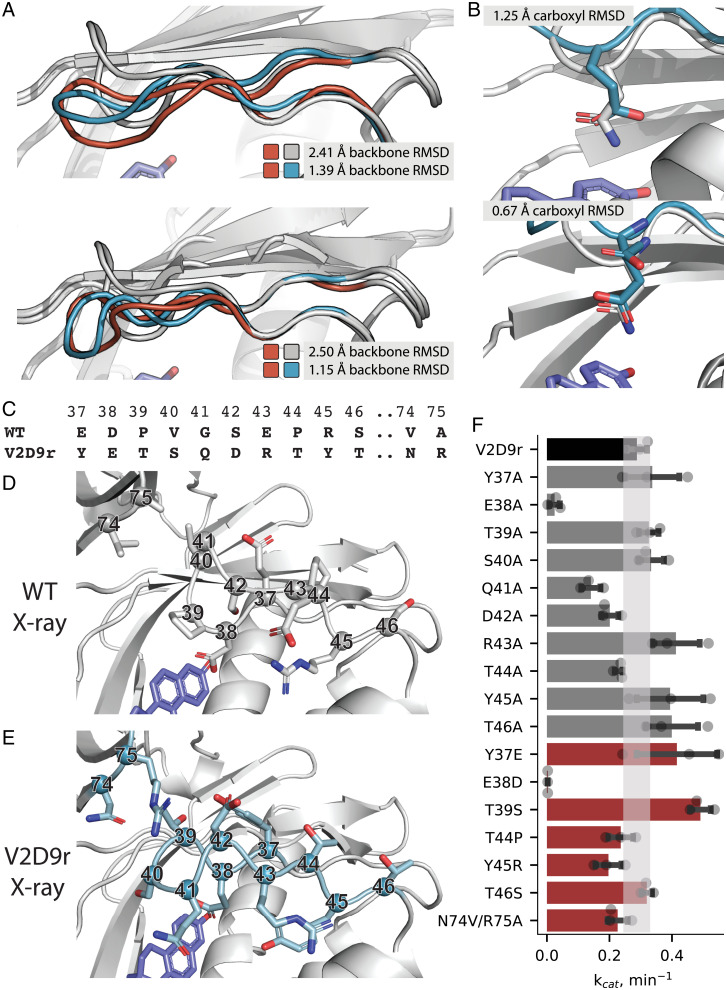
SignificanceComputational protein design promises to advance applications in medicine and biotechnology by creating proteins with many new and useful functions. However, new functions require the design of specific and often irregular atom-level geometries, which remains a major challenge. Here, we develop computational methods that design and predict local protein geometries with greater accuracy than existing methods. Then, as a proof of concept, we leverage these methods to design new protein conformations in the enzyme ketosteroid isomerase that change the protein's preference for a key functional residue. Our computational methods are openly accessible and can be applied to the design of other intricate geometries customized for new user-defined protein functions.
Keywords: Rosetta; computational protein design; design of function; structure prediction.
Conflict of interest statement
Competing interest statement: J.J.G. and T.K. are unpaid board members of the Rosetta Commons.
Figures