

β-catenin regulates HIV latency and modulates HIV reactivation
- PMID: 35255110
- PMCID: PMC8939789
- DOI: 10.1371/journal.ppat.1010354
β-catenin regulates HIV latency and modulates HIV reactivation
Abstract
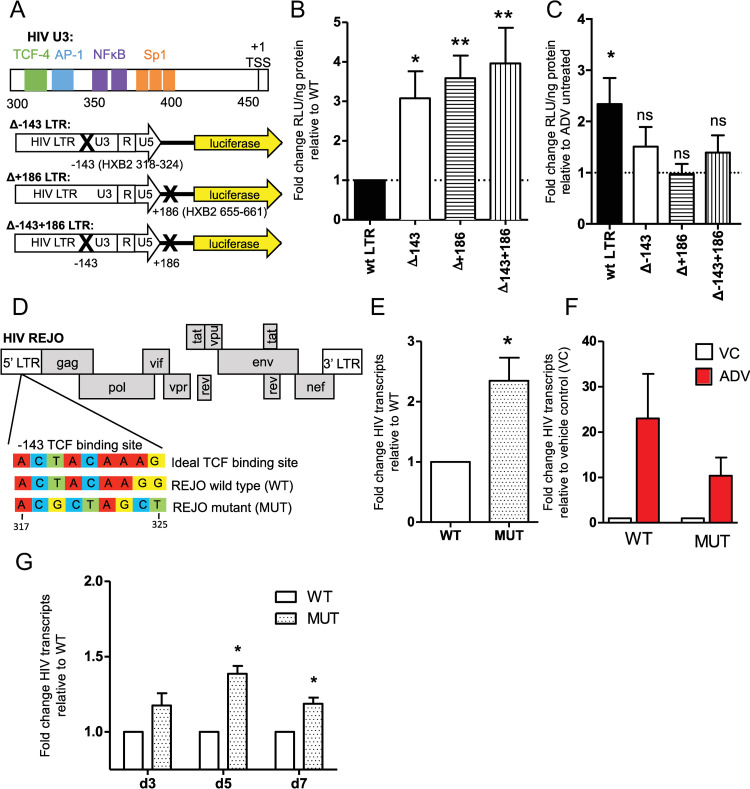
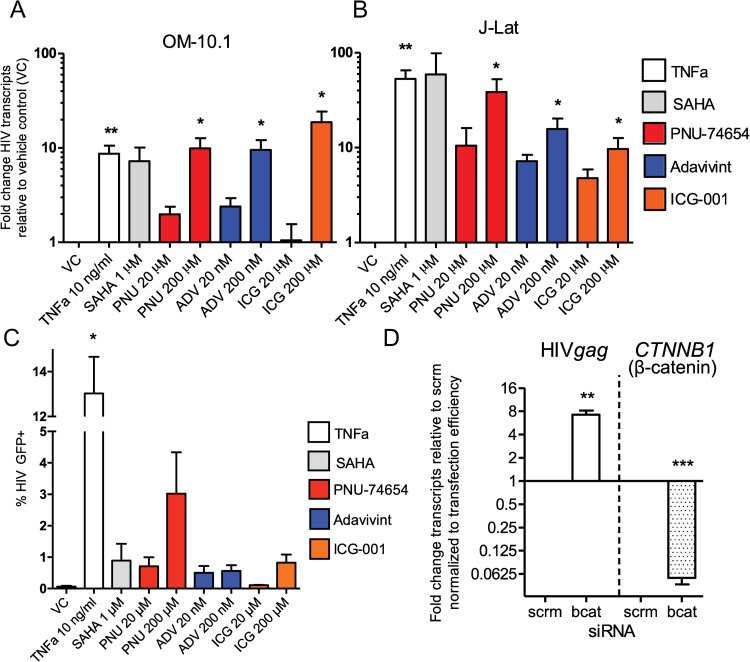
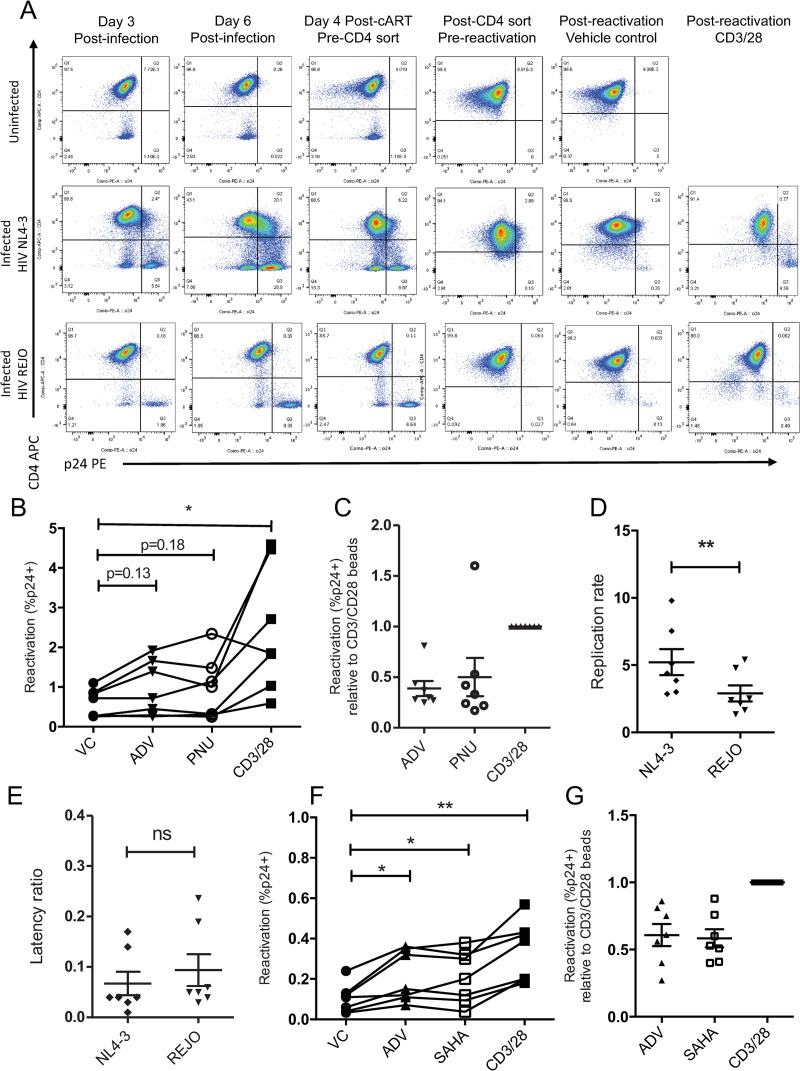
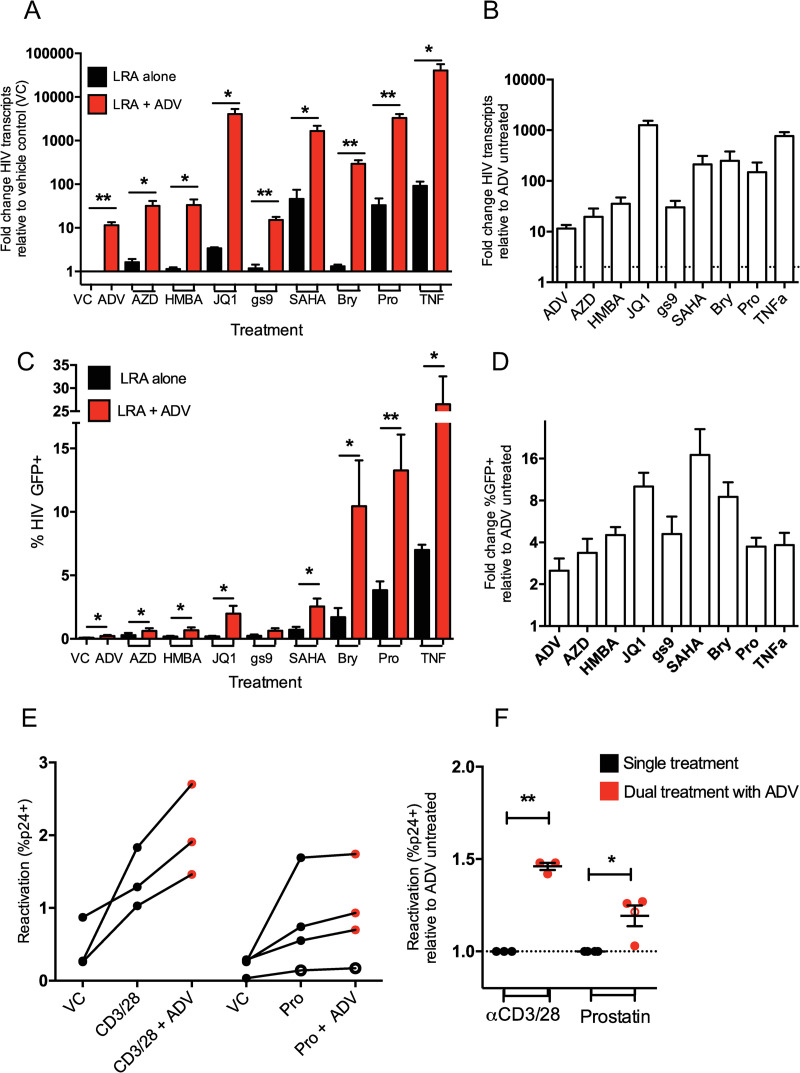
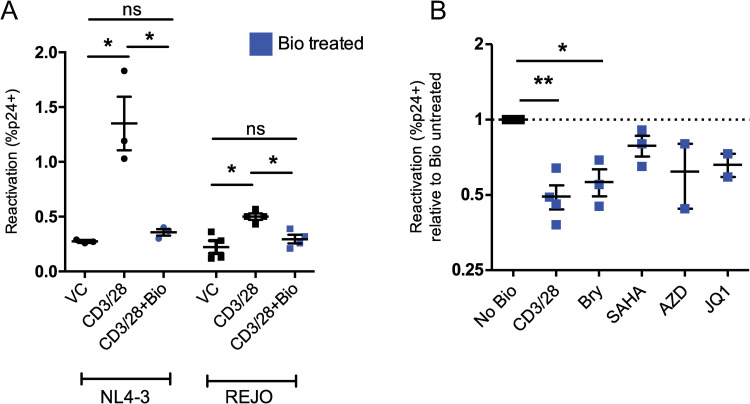
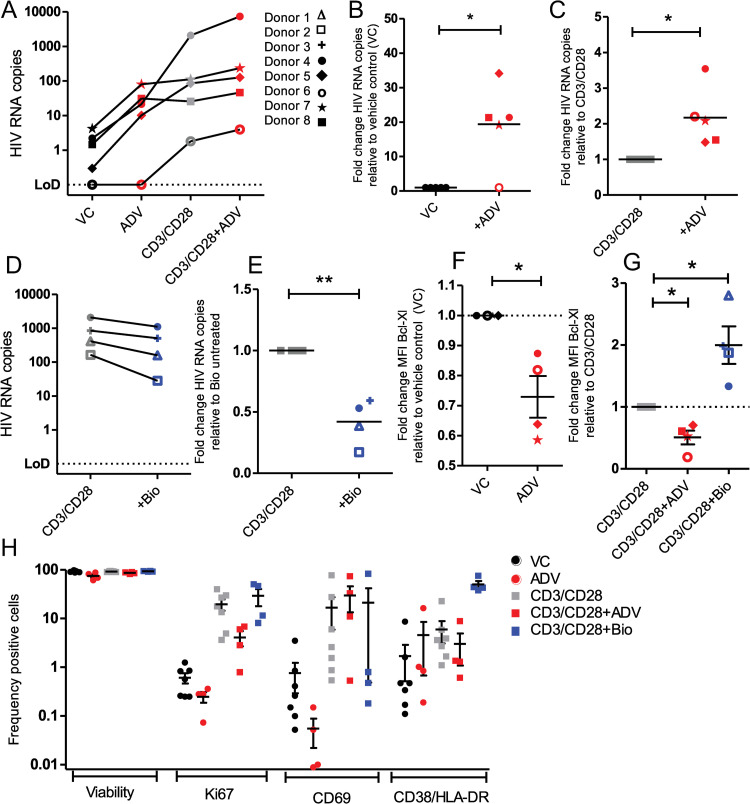
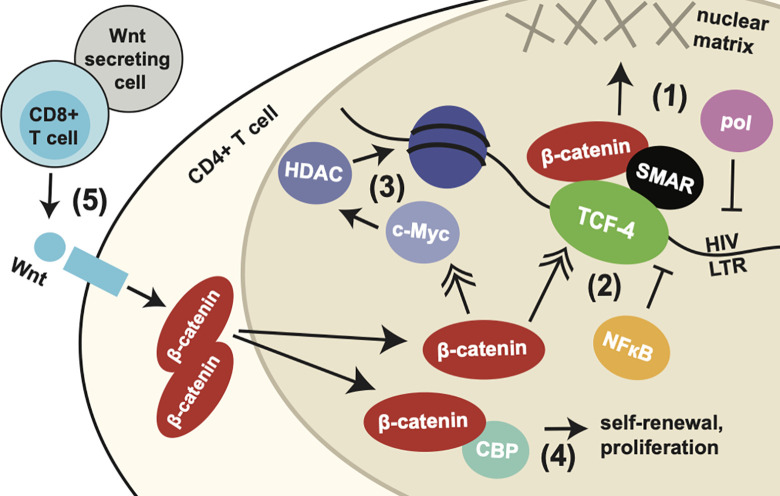
Latency is the main obstacle towards an HIV cure, with cure strategies aiming to either elicit or prevent viral reactivation. While these strategies have shown promise, they have only succeeded in modulating latency in a fraction of the latent HIV reservoir, suggesting that the mechanisms controlling HIV latency are not completely understood, and that comprehensive latency modulation will require targeting of multiple latency maintenance pathways. We show here that the transcriptional co-activator and the central mediator of canonical Wnt signaling, β-catenin, inhibits HIV transcription in CD4+ T cells via TCF-4 LTR binding sites. Further, we show that inhibiting the β-catenin pathway reactivates HIV in a primary TCM cell model of HIV latency, primary cells from cART-controlled HIV donors, and in CD4+ latent cell lines. β-catenin inhibition or activation also enhanced or inhibited the activity of several classes of HIV latency reversing agents, respectively, in these models, with significant synergy of β-catenin and each LRA class tested. In sum, we identify β-catenin as a novel regulator of HIV latency in vitro and ex vivo, adding new therapeutic targets that may be combined for comprehensive HIV latency modulation in HIV cure efforts.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
References
-
- Joseph SB, Kincer LP, Bowman NM, Evans C, Vinikoor MJ, Lippincott CK, et al.. Human Immunodeficiency Virus Type 1 RNA Detected in the Central Nervous System (CNS) After Years of Suppressive Antiretroviral Therapy Can Originate from a Replicating CNS Reservoir or Clonally Expanded Cells. Clinical Infectious Diseases. 2019;69(8):1345–52. doi: 10.1093/cid/ciy1066 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
