

StrainGE: a toolkit to track and characterize low-abundance strains in complex microbial communities
- PMID: 35255937
- PMCID: PMC8900328
- DOI: 10.1186/s13059-022-02630-0
StrainGE: a toolkit to track and characterize low-abundance strains in complex microbial communities
Abstract
Human-associated microbial communities comprise not only complex mixtures of bacterial species, but also mixtures of conspecific strains, the implications of which are mostly unknown since strain level dynamics are underexplored due to the difficulties of studying them. We introduce the Strain Genome Explorer (StrainGE) toolkit, which deconvolves strain mixtures and characterizes component strains at the nucleotide level from short-read metagenomic sequencing with higher sensitivity and resolution than other tools. StrainGE is able to identify strains at 0.1x coverage and detect variants for multiple conspecific strains within a sample from coverages as low as 0.5x.
Keywords: Metagenomics; Microbiome; Strain-tracking.
© 2022. The Author(s).
Conflict of interest statement
BJW is an employee of Applied Invention (Cambridge, MA). No other authors declare competing interests.
Figures
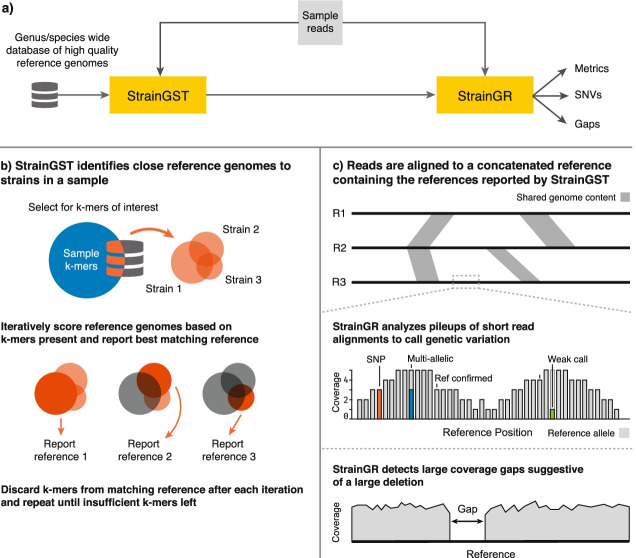

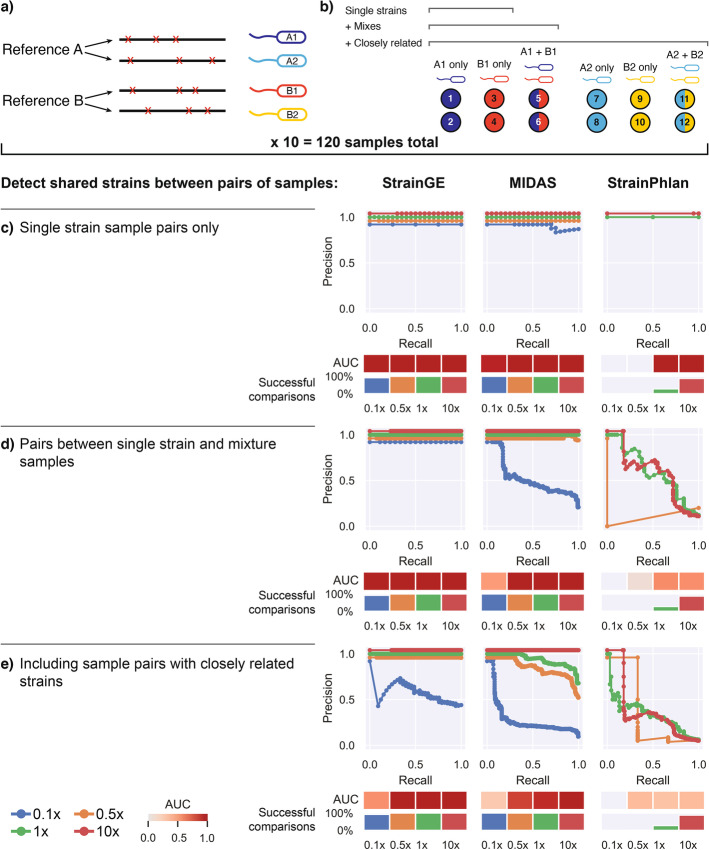

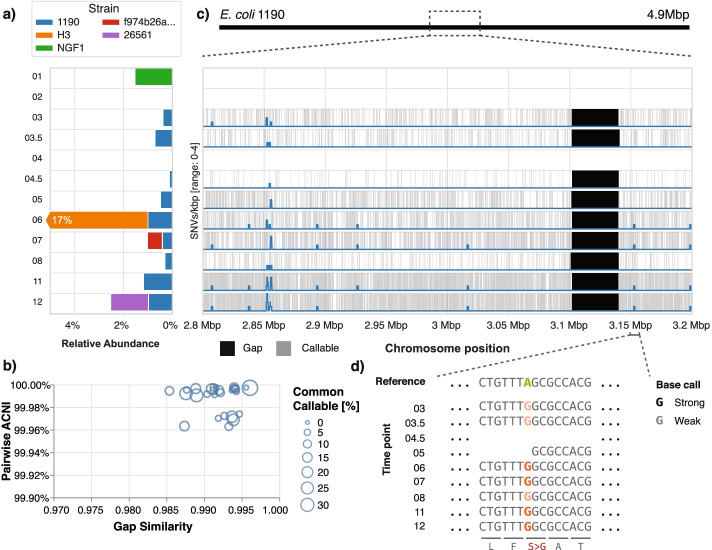

References
-
- Leimbach A, Hacker J, Dobrindt U. E. coli as an All-Rounder: the thin line between commensalism and pathogenicity. In: Dobrindt U, Hacker JH, Svanborg C, editors. Between pathogenicity and commensalism. Berlin, Heidelberg: Springer; 2013. pp. 3–32. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
