

Novel approaches to diagnosis and management of hereditary transthyretin amyloidosis
- PMID: 35256455
- PMCID: PMC9148983
- DOI: 10.1136/jnnp-2021-327909
Novel approaches to diagnosis and management of hereditary transthyretin amyloidosis
Abstract
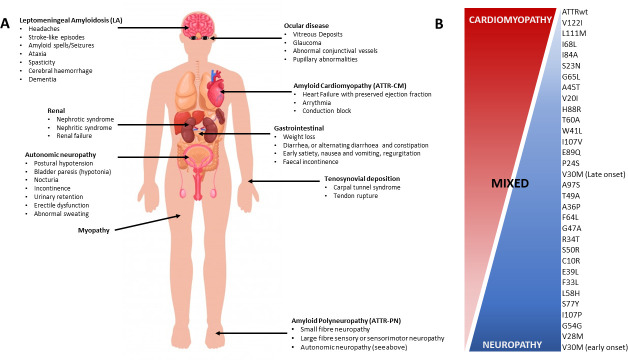
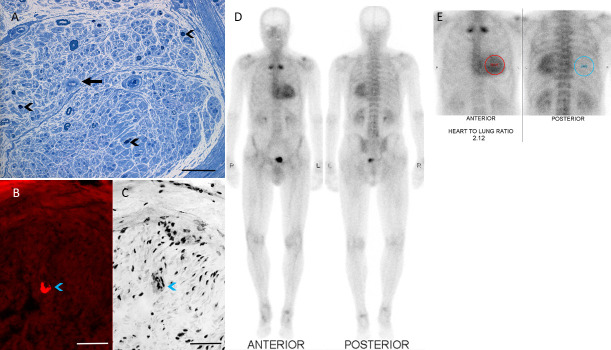
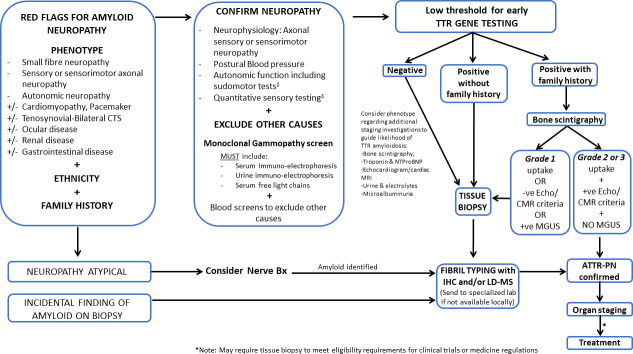
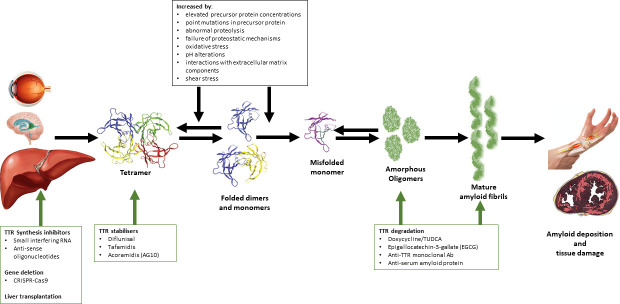
Hereditary transthyretin amyloidosis (ATTRv) is a severe, adult-onset autosomal dominant inherited systemic disease predominantly affecting the peripheral and autonomic nervous system, heart, kidney and the eyes. ATTRv is caused by mutations of the transthyretin (TTR) gene, leading to extracellular deposition of amyloid fibrils in multiple organs including the peripheral nervous system. Typically, the neuropathy associated with ATTRv is characterised by a rapidly progressive and disabling sensorimotor axonal neuropathy with early small-fibre involvement. Carpal tunnel syndrome and cardiac dysfunction frequently coexist as part of the ATTRv phenotype. Although awareness of ATTRv polyneuropathy among neurologists has increased, the rate of misdiagnosis remains high, resulting in significant diagnostic delays and accrued disability. A timely and definitive diagnosis is important, given the emergence of effective therapies which have revolutionised the management of transthyretin amyloidosis. TTR protein stabilisers diflunisal and tafamidis can delay the progression of the disease, if treated early in the course. Additionally, TTR gene silencing medications, patisiran and inotersen, have resulted in up to 80% reduction in TTR production, leading to stabilisation or slight improvement of peripheral neuropathy and cardiac dysfunction, as well as improvement in quality of life and functional outcomes. The considerable therapeutic advances have raised additional challenges, including optimisation of diagnostic techniques and management approaches in ATTRv neuropathy. This review highlights the key advances in the diagnostic techniques, current and emerging management strategies, and biomarker development for disease progression in ATTRv.
Keywords: amyloid; neuropathy.
© Author(s) (or their employer(s)) 2022. Re-use permitted under CC BY-NC. No commercial re-use. See rights and permissions. Published by BMJ.
Conflict of interest statement
Competing interests: Author MMR has consulted for Alnylam, IONIS and AKCEA.
Figures
References
Publication types
MeSH terms
Substances
Supplementary concepts
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous