

An anti-PD-1-GITR-L bispecific agonist induces GITR clustering-mediated T cell activation for cancer immunotherapy
- PMID: 35256819
- PMCID: PMC8960412
- DOI: 10.1038/s43018-022-00334-9
An anti-PD-1-GITR-L bispecific agonist induces GITR clustering-mediated T cell activation for cancer immunotherapy
Abstract
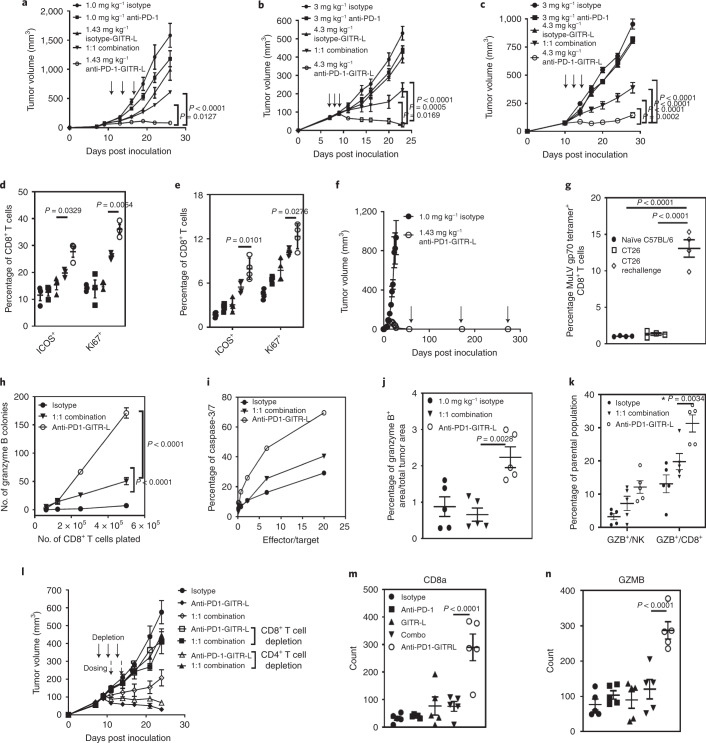
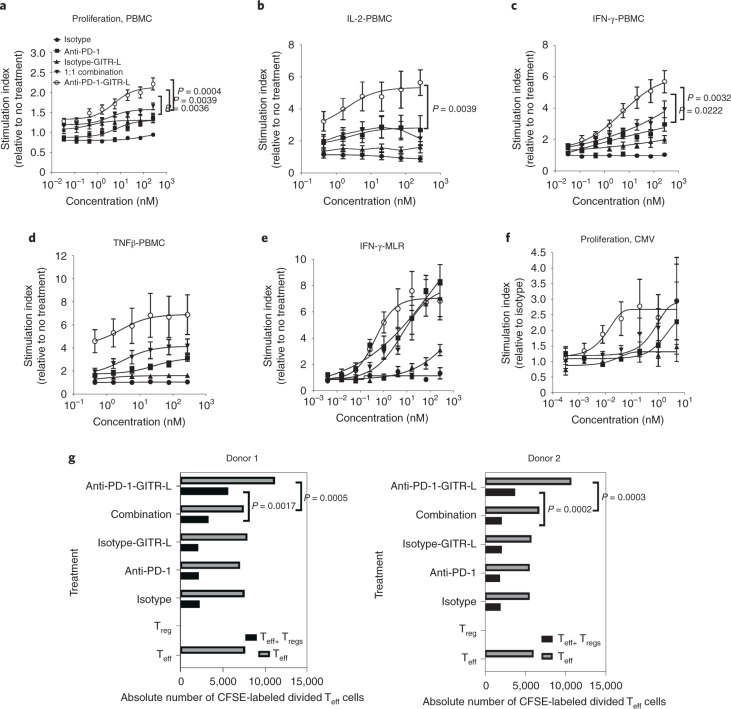
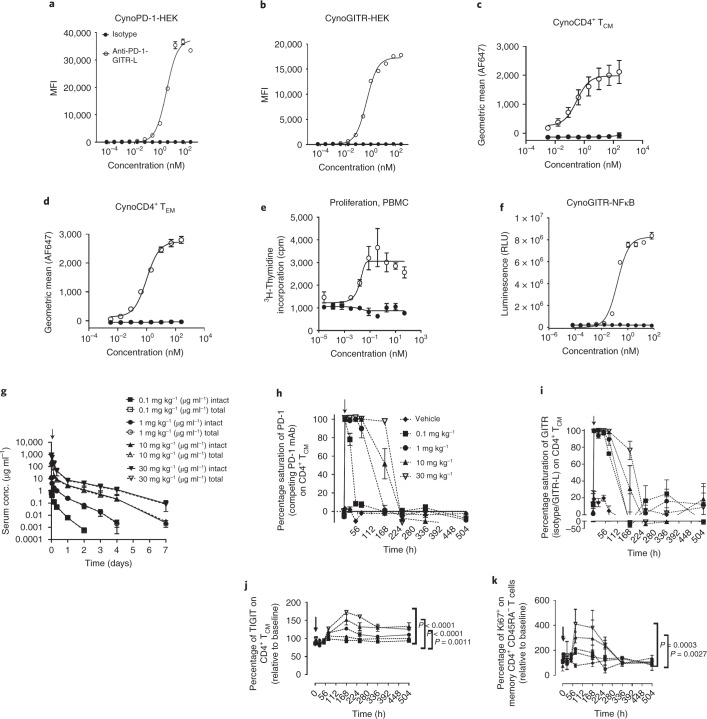
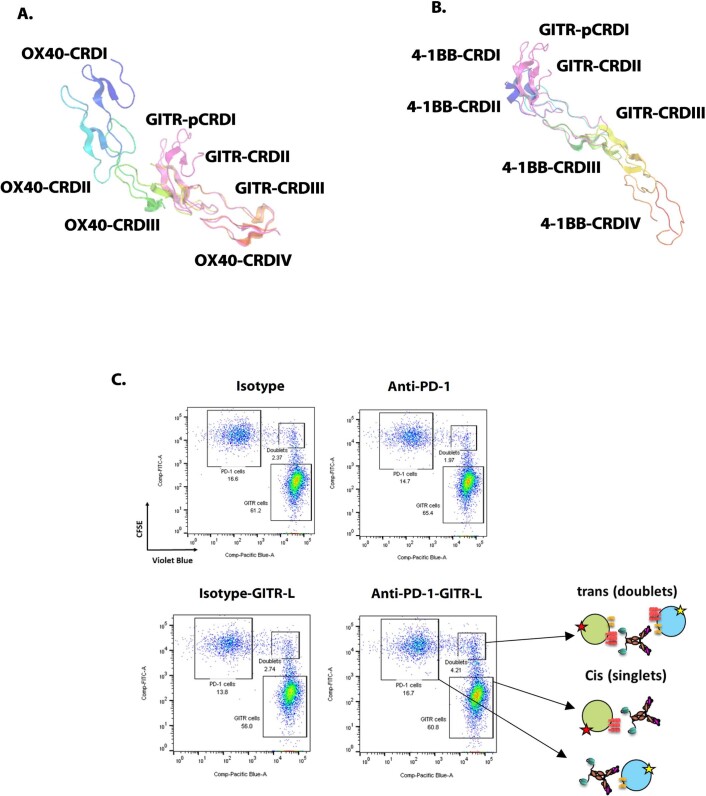
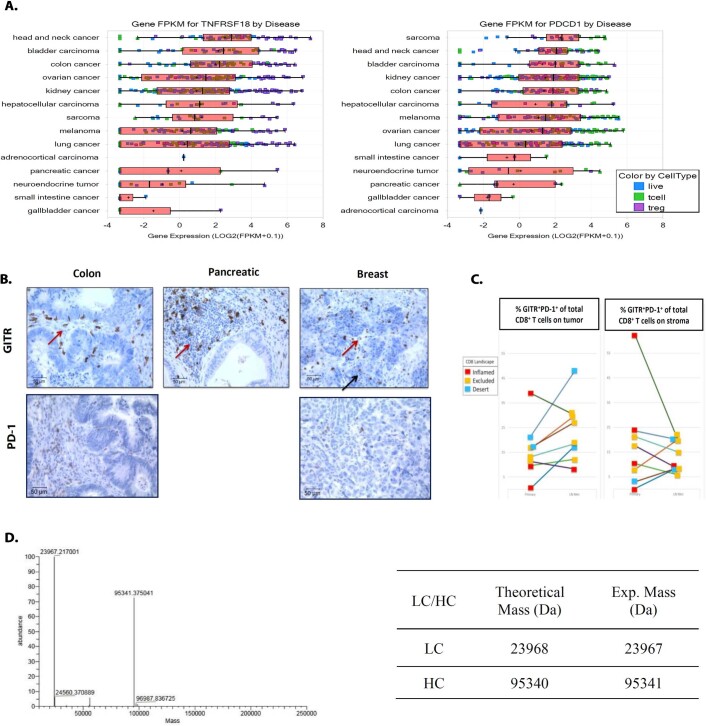
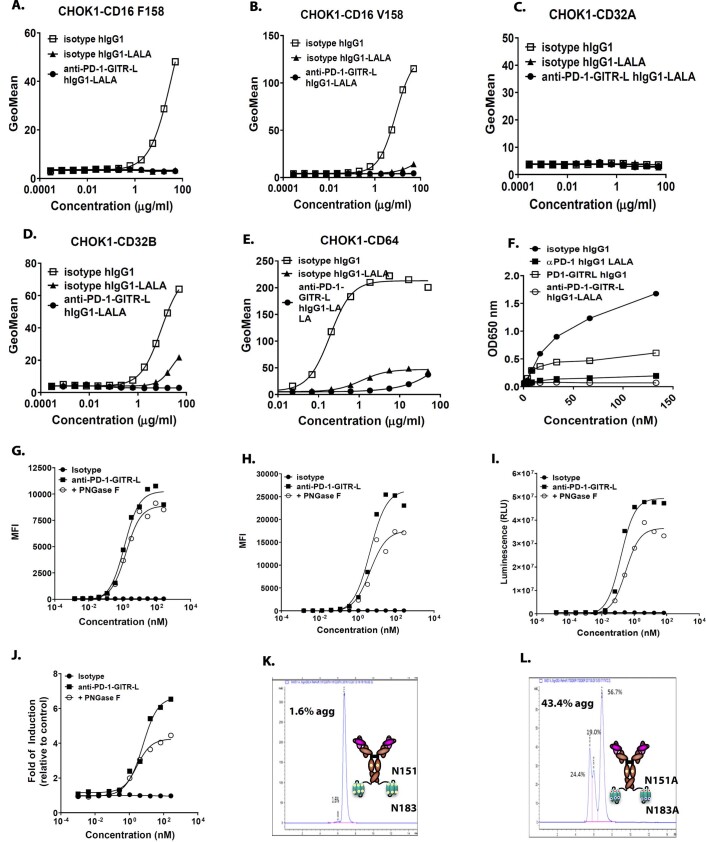
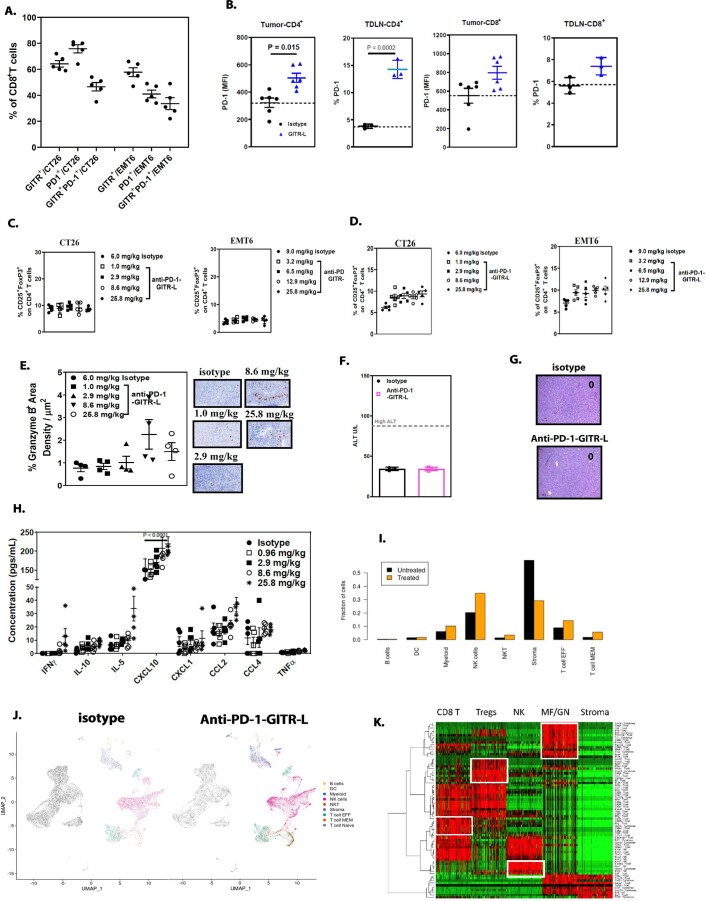
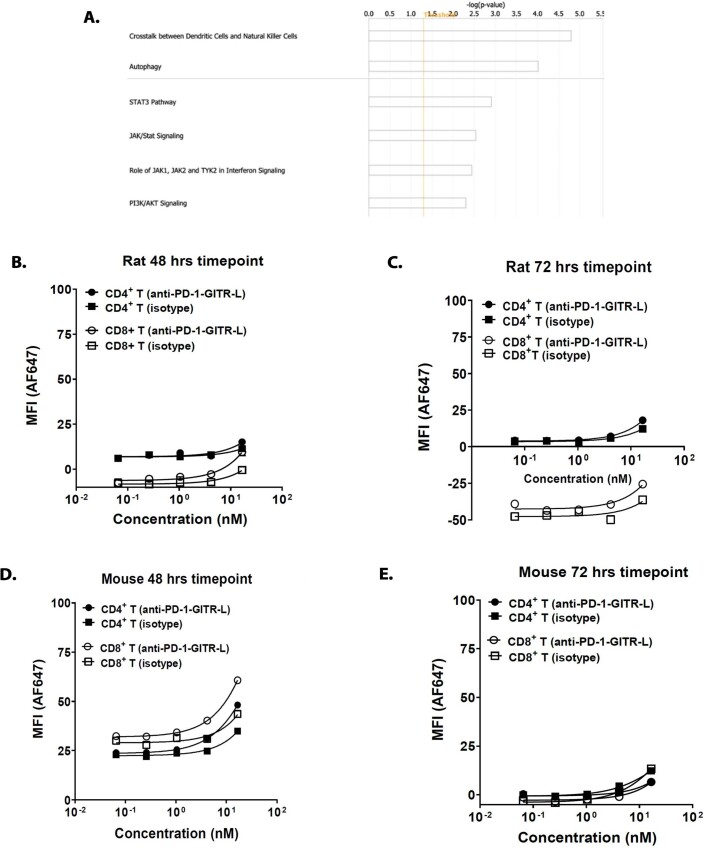
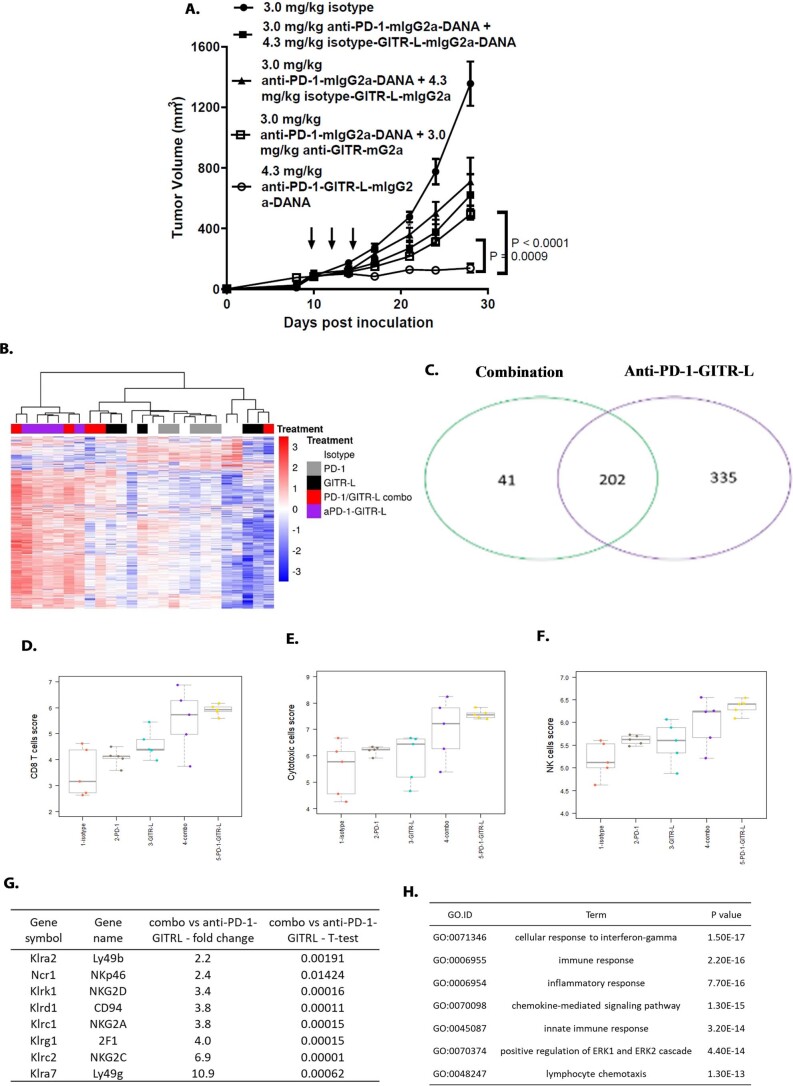
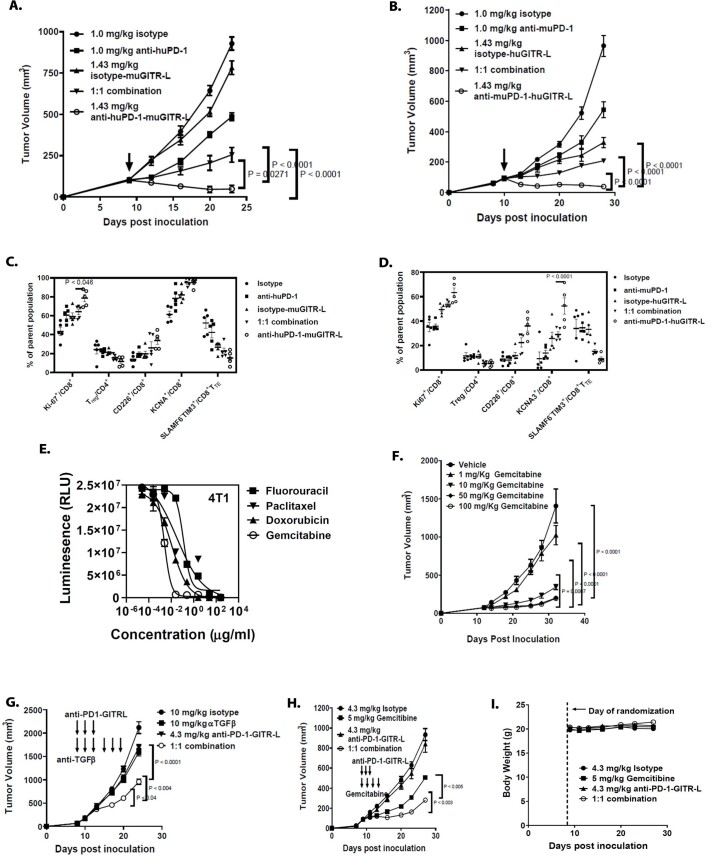
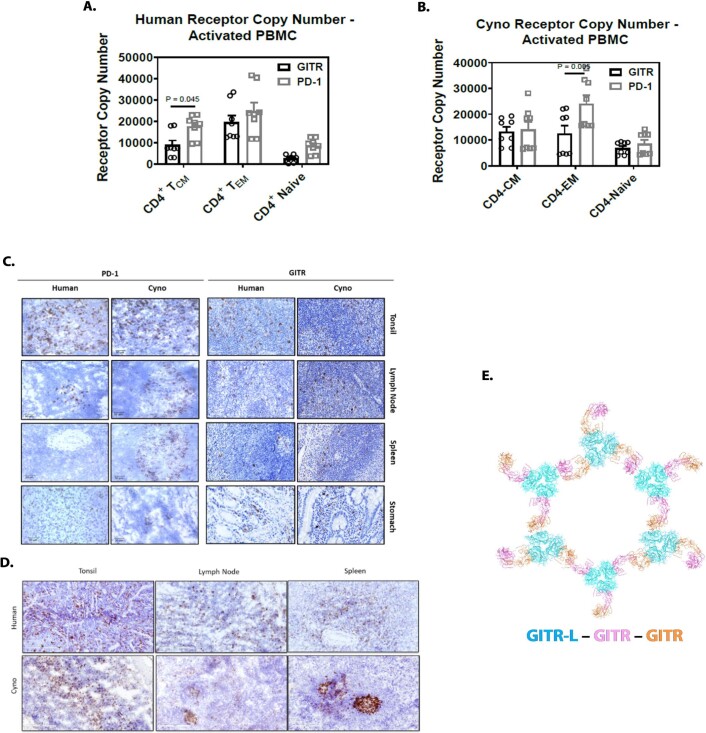
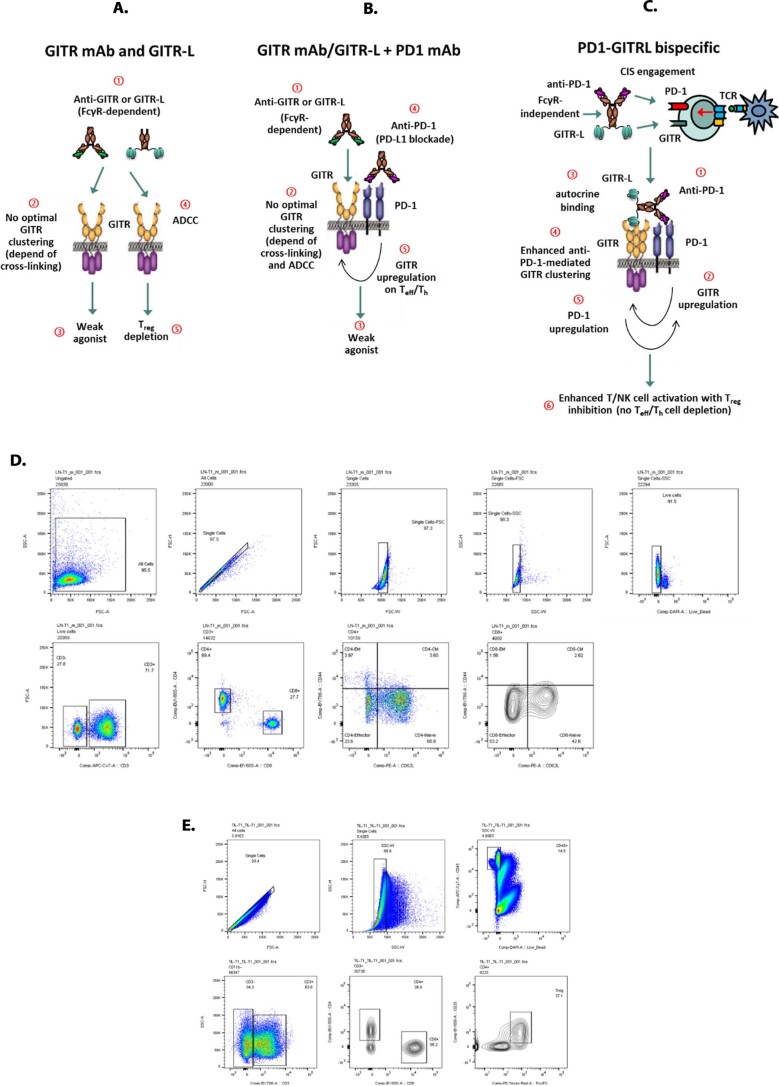
Costimulatory receptors such as glucocorticoid-induced tumor necrosis factor receptor-related protein (GITR) play key roles in regulating the effector functions of T cells. In human clinical trials, however, GITR agonist antibodies have shown limited therapeutic effect, which may be due to suboptimal receptor clustering-mediated signaling. To overcome this potential limitation, a rational protein engineering approach is needed to optimize GITR agonist-based immunotherapies. Here we show a bispecific molecule consisting of an anti-PD-1 antibody fused with a multimeric GITR ligand (GITR-L) that induces PD-1-dependent and FcγR-independent GITR clustering, resulting in enhanced activation, proliferation and memory differentiation of primed antigen-specific GITR+PD-1+ T cells. The anti-PD-1-GITR-L bispecific is a PD-1-directed GITR-L construct that demonstrated dose-dependent, immunologically driven tumor growth inhibition in syngeneic, genetically engineered and xenograft humanized mouse tumor models, with a dose-dependent correlation between target saturation and Ki67 and TIGIT upregulation on memory T cells. Anti-PD-1-GITR-L thus represents a bispecific approach to directing GITR agonism for cancer immunotherapy.
© 2022. The Author(s).
Conflict of interest statement
S.C., N.B., S.H., B.R., M.G., E.L., N.T., D.Z., M.S., W.L., P.Z., Z.D., K.H., S.R., A.T., C.F., M.X., S.T., R.I., M.L., E.D., L.Z., L.B., K.L., J.P.R., C.L., M.T., M.F., Y.A. and H.M.A. are employees of AbbVie. M.S., P.G., T.M., A.W., H.K., H.Z., M.C., B.C., D.H. and F.H. were employees of AbbVie at the time of the study. The design, study conduct and financial support for this research were provided by AbbVie. AbbVie participated in the interpretation of data and review and approval of the publication. J.P.R. declares no competing interests.
Figures


Comment in
-
Bispecific agonist boosts anti-tumour T cells via GITR.Nat Rev Immunol. 2022 Apr;22(4):208. doi: 10.1038/s41577-022-00708-1. Nat Rev Immunol. 2022. PMID: 35296813 No abstract available.
Similar articles
-
Rational design of anti-GITR-based combination immunotherapy.Nat Med. 2019 May;25(5):759-766. doi: 10.1038/s41591-019-0420-8. Epub 2019 Apr 29. Nat Med. 2019. PMID: 31036879 Free PMC article. Clinical Trial.
-
Combined PD-1 blockade and GITR triggering induce a potent antitumor immunity in murine cancer models and synergizes with chemotherapeutic drugs.J Transl Med. 2014 Feb 7;12:36. doi: 10.1186/1479-5876-12-36. J Transl Med. 2014. PMID: 24502656 Free PMC article.
-
HERA-GITRL activates T cells and promotes anti-tumor efficacy independent of FcγR-binding functionality.J Immunother Cancer. 2019 Jul 19;7(1):191. doi: 10.1186/s40425-019-0671-4. J Immunother Cancer. 2019. PMID: 31324216 Free PMC article.
-
T-cell intrinsic effects of GITR and 4-1BB during viral infection and cancer immunotherapy.Immunol Rev. 2011 Nov;244(1):197-217. doi: 10.1111/j.1600-065X.2011.01063.x. Immunol Rev. 2011. PMID: 22017440 Review.
-
Rationale for anti-GITR cancer immunotherapy.Eur J Cancer. 2016 Nov;67:1-10. doi: 10.1016/j.ejca.2016.06.028. Epub 2016 Aug 31. Eur J Cancer. 2016. PMID: 27591414 Review.
Cited by
-
Mitogen-Activated Protein Kinase and Nuclear Hormone Receptor Crosstalk in Cancer Immunotherapy.Int J Mol Sci. 2023 Sep 4;24(17):13661. doi: 10.3390/ijms241713661. Int J Mol Sci. 2023. PMID: 37686465 Free PMC article. Review.
-
Targeting GITR in cancer immunotherapy - there is no perfect knowledge.Oncotarget. 2023 Jun 19;14:614-621. doi: 10.18632/oncotarget.28461. Oncotarget. 2023. PMID: 37335294 Free PMC article.
-
Emerging IO checkpoints in gastrointestinal oncology.Front Immunol. 2025 Jul 24;16:1575713. doi: 10.3389/fimmu.2025.1575713. eCollection 2025. Front Immunol. 2025. PMID: 40777027 Free PMC article. Review.
-
Transforming growth factor-β signalling in tumour resistance to the anti-PD-(L)1 therapy: Updated.J Cell Mol Med. 2023 Feb;27(3):311-321. doi: 10.1111/jcmm.17666. Epub 2023 Jan 10. J Cell Mol Med. 2023. PMID: 36625080 Free PMC article. Review.
-
Development of pharmacological immunoregulatory anti-cancer therapeutics: current mechanistic studies and clinical opportunities.Signal Transduct Target Ther. 2024 May 22;9(1):126. doi: 10.1038/s41392-024-01826-z. Signal Transduct Target Ther. 2024. PMID: 38773064 Free PMC article. Review.
References
-
- Sharpe AH, Pauken KE. The diverse functions of the PD1 inhibitory pathway. Nat. Rev. Immunol. 2017;18:153–167. - PubMed
-
- Wei SC, Duffy DR, Allison JP. Fundamental mechanism of immune checkpoint blockade therapy. Cancer Discov. 2018;8:1069–1086. - PubMed
-
- Kim JM, Chen DS. Immune escape to PD-L1/PD-1 blockade: seven steps to success (or failure) Ann. Oncol. 2016;27:1492–1504. - PubMed
-
- Clouthier DL, Watts TH. Cell-specific and context-dependent effects of GITR in cancer, autoimmunity, and infection. Cytokine Growth Factor Rev. 2014;25:91–106. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
