

Targeting whole body metabolism and mitochondrial bioenergetics in the drug development for Alzheimer's disease
- PMID: 35256932
- PMCID: PMC8897048
- DOI: 10.1016/j.apsb.2021.06.014
Targeting whole body metabolism and mitochondrial bioenergetics in the drug development for Alzheimer's disease
Abstract
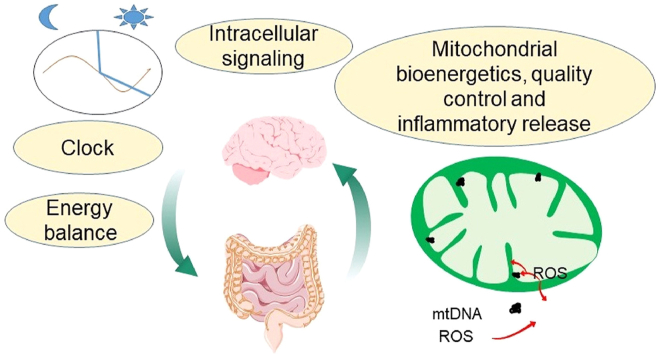
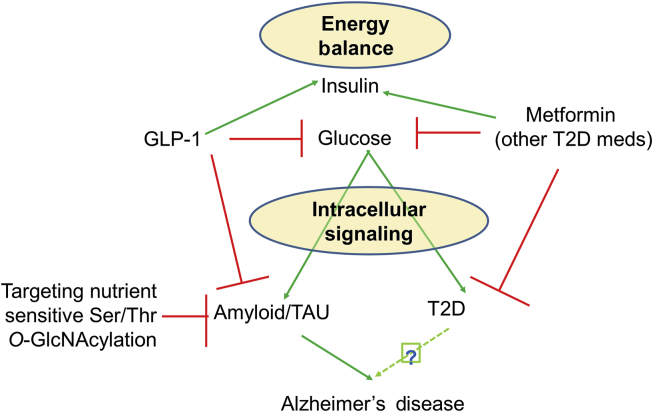
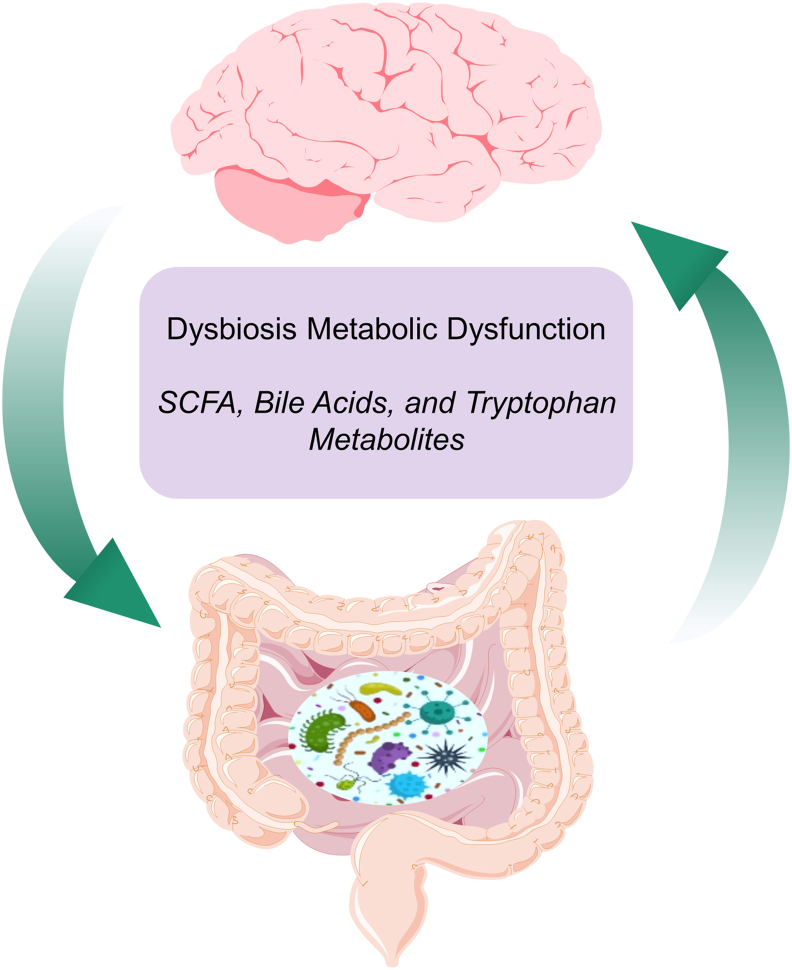
Aging is by far the most prominent risk factor for Alzheimer's disease (AD), and both aging and AD are associated with apparent metabolic alterations. As developing effective therapeutic interventions to treat AD is clearly in urgent need, the impact of modulating whole-body and intracellular metabolism in preclinical models and in human patients, on disease pathogenesis, have been explored. There is also an increasing awareness of differential risk and potential targeting strategies related to biological sex, microbiome, and circadian regulation. As a major part of intracellular metabolism, mitochondrial bioenergetics, mitochondrial quality-control mechanisms, and mitochondria-linked inflammatory responses have been considered for AD therapeutic interventions. This review summarizes and highlights these efforts.
Keywords: ACE2, angiotensin I converting enzyme (peptidyl-dipeptidase A) 2; AD, Alzheimer's disease; ADP, adenosine diphosphate; ADRD, AD-related dementias; Aβ, amyloid β; CSF, cerebrospinal fluid; Circadian regulation; DAMPs; DAMPs, damage-associated molecular patterns; Diabetes; ER, estrogen receptor; ETC, electron transport chain; FCCP, trifluoromethoxy carbonylcyanide phenylhydrazone; FPR-1, formyl peptide receptor 1; GIP, glucose-dependent insulinotropic polypeptide; GLP-1, glucagon-like peptide-1; HBP, hexoamine biosynthesis pathway; HTRA, high temperature requirement A; Hexokinase biosynthesis pathway; I3A, indole-3-carboxaldehyde; IRF-3, interferon regulatory factor 3; LC3, microtubule associated protein light chain 3; LPS, lipopolysaccharide; LRR, leucine-rich repeat; MAVS, mitochondrial anti-viral signaling; MCI, mild cognitive impairment; MRI, magnetic resonance imaging; MRS, magnetic resonance spectroscopy; Mdivi-1, mitochondrial division inhibitor 1; Microbiome; Mitochondrial DNA; Mitochondrial electron transport chain; Mitochondrial quality control; NLRP3, leucine-rich repeat (LRR)-containing protein (NLR)-like receptor family pyrin domain containing 3; NOD, nucleotide-binding oligomerization domain; NeuN, neuronal nuclear protein; PET, fluorodeoxyglucose (FDG)-positron emission tomography; PKA, protein kinase A; POLβ, the base-excision repair enzyme DNA polymerase β; ROS, reactive oxygen species; Reactive species; SAMP8, senescence-accelerated mice; SCFAs, short-chain fatty acids; SIRT3, NAD-dependent deacetylase sirtuin-3; STING, stimulator of interferon genes; STZ, streptozotocin; SkQ1, plastoquinonyldecyltriphenylphosphonium; T2D, type 2 diabetes; TCA, Tricarboxylic acid; TLR9, toll-like receptor 9; TMAO, trimethylamine N-oxide; TP, tricyclic pyrone; TRF, time-restricted feeding; cAMP, cyclic adenosine monophosphate; cGAS, cyclic GMP/AMP synthase; hAPP, human amyloid precursor protein; hPREP, human presequence protease; i.p., intraperitoneal; mTOR, mechanistic target of rapamycin; mtDNA, mitochondrial DNA; αkG, alpha-ketoglutarate.
© 2022 Chinese Pharmaceutical Association and Institute of Materia Medica, Chinese Academy of Medical Sciences. Production and hosting by Elsevier B.V.
Figures
References
-
- 2020 Alzheimer’s disease facts and figures. Alzheimers Dement. 2020;16:391–460.
-
- 2021 Alzheimer’s disease facts and figures. Alzheimers Dement. 2021;17:327–406. - PubMed
-
- Murphy S.L., Xu J., Kochanek K.D., Arias E., Tejada-Vera B. Deaths: final data for 2018. Natl Vital Stat Rep. 2021;69:1–83. - PubMed
-
- Hoyer S. Glucose metabolism and insulin receptor signal transduction in Alzheimer disease. Eur J Pharmacol. 2004;490:115–125. - PubMed
-
- Cheng G., Huang C., Deng H., Wang H. Diabetes as a risk factor for dementia and mild cognitive impairment: a meta-analysis of longitudinal studies. Intern Med J. 2012;42:484–491. - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
