

Mitochondrial respiration supports autophagy to provide stress resistance during quiescence
- PMID: 35258392
- PMCID: PMC9542673
- DOI: 10.1080/15548627.2022.2038898
Mitochondrial respiration supports autophagy to provide stress resistance during quiescence
Abstract
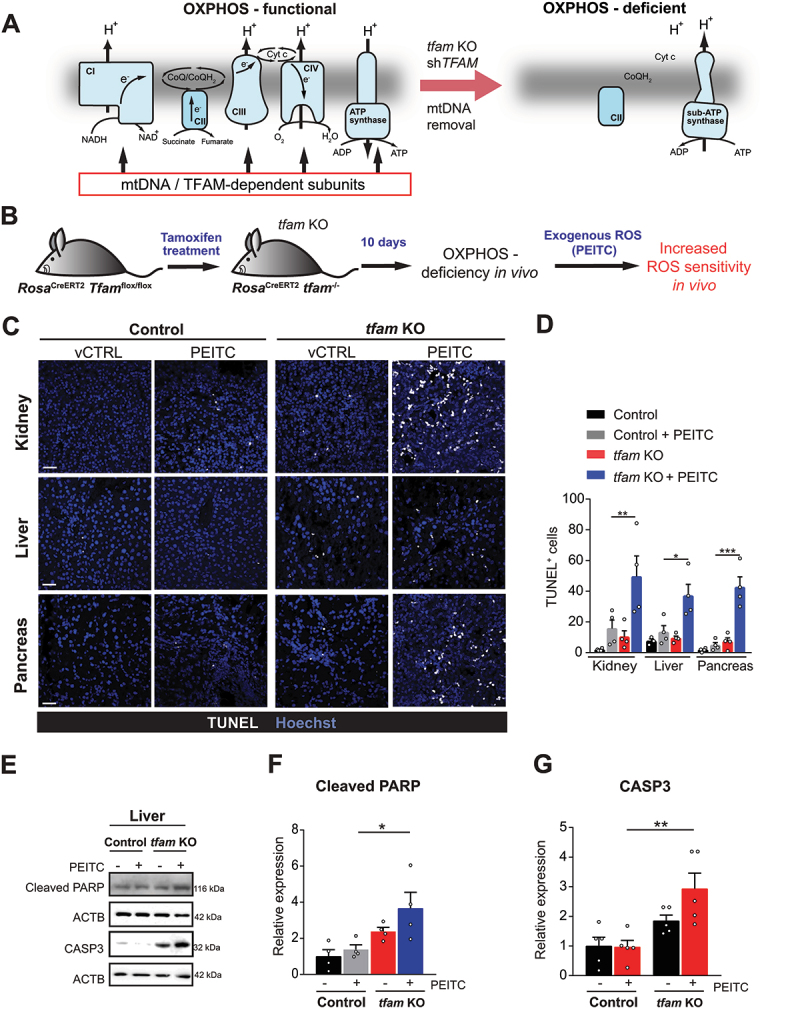
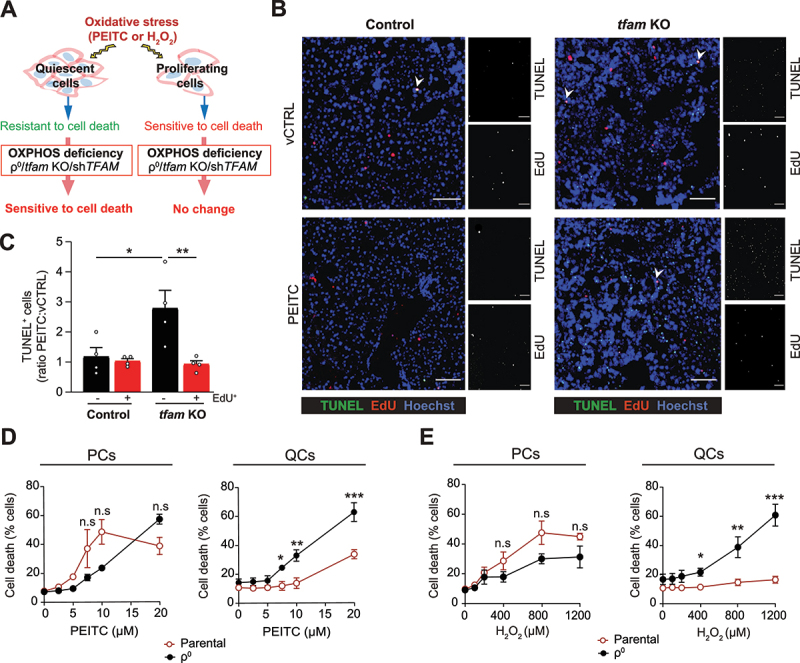
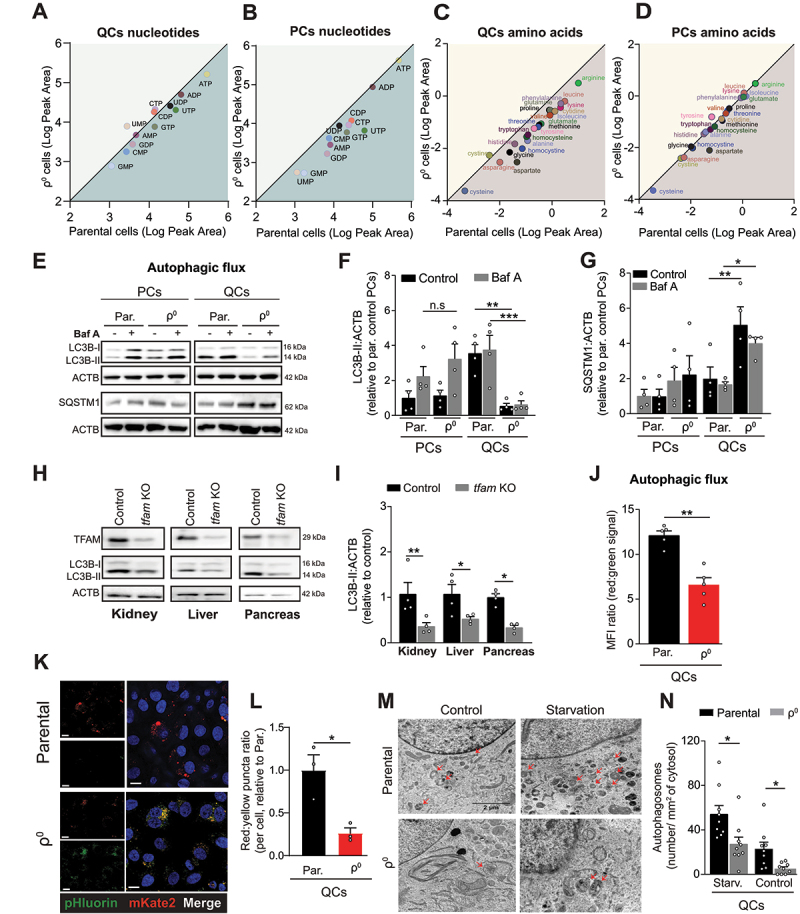
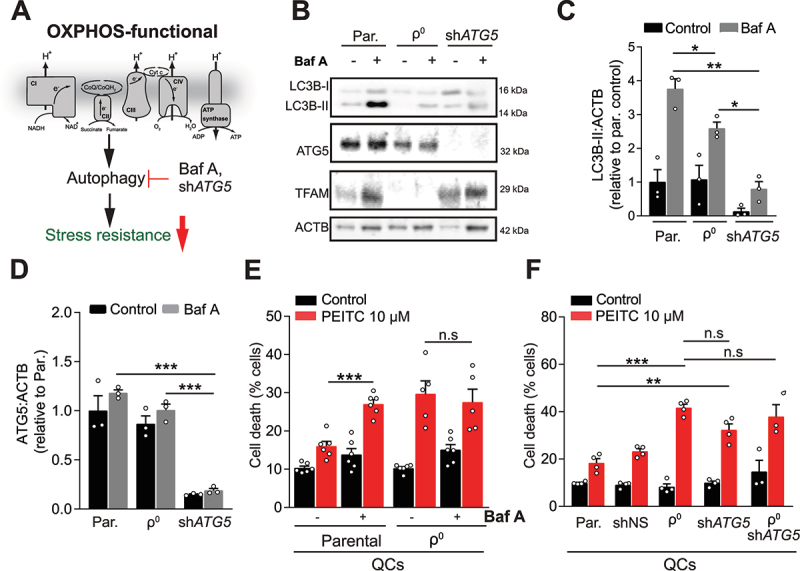
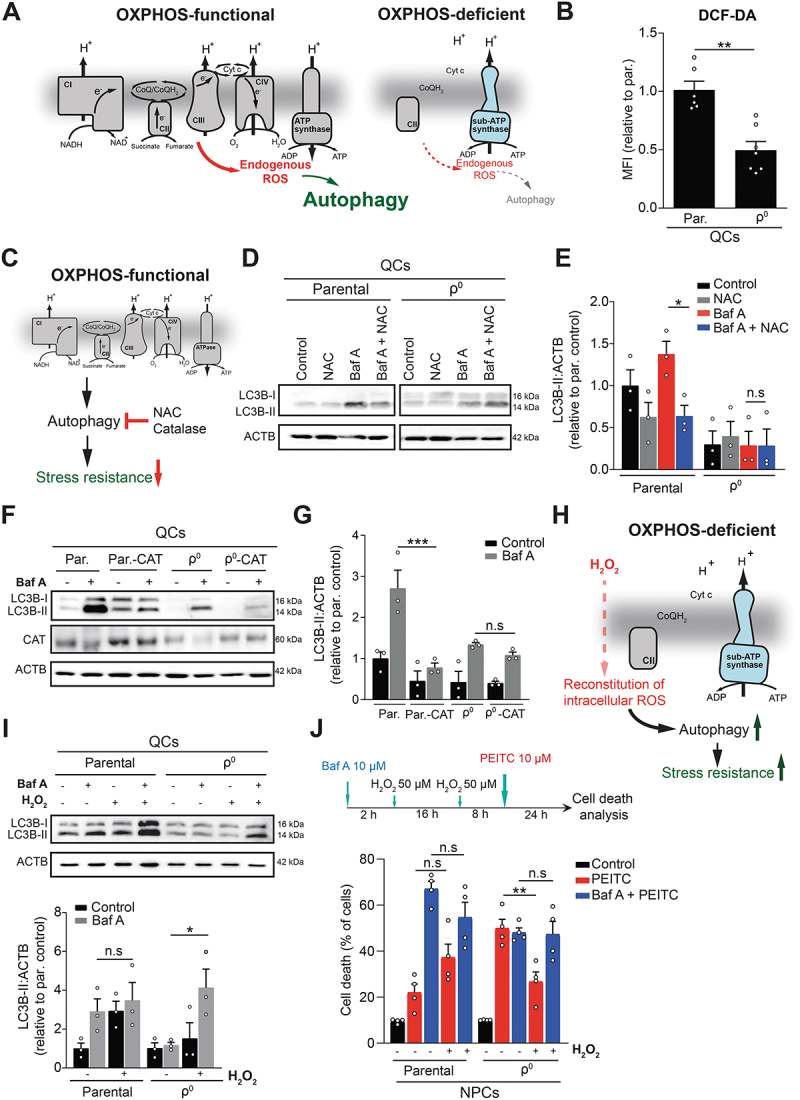
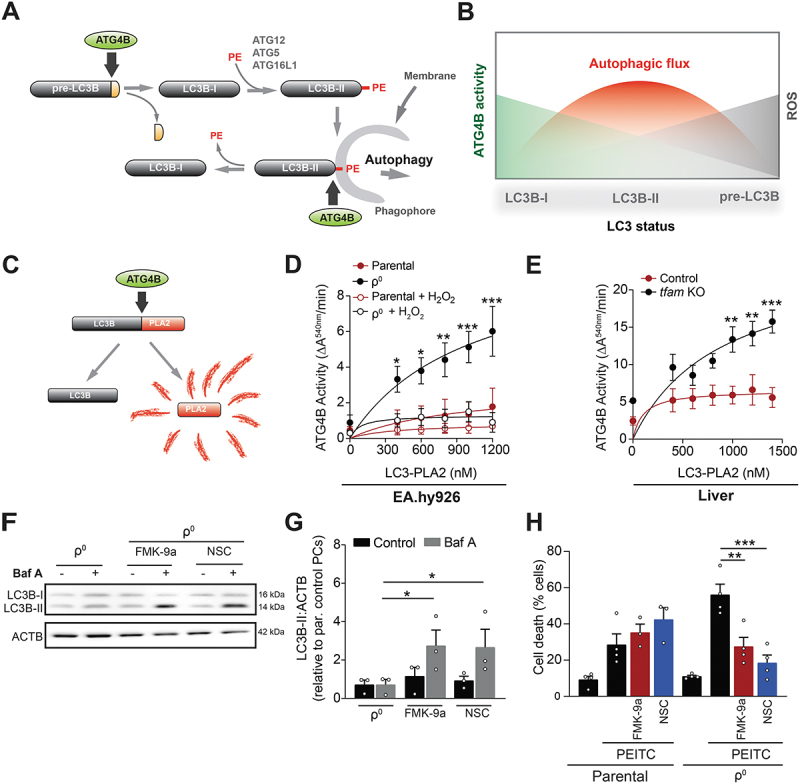
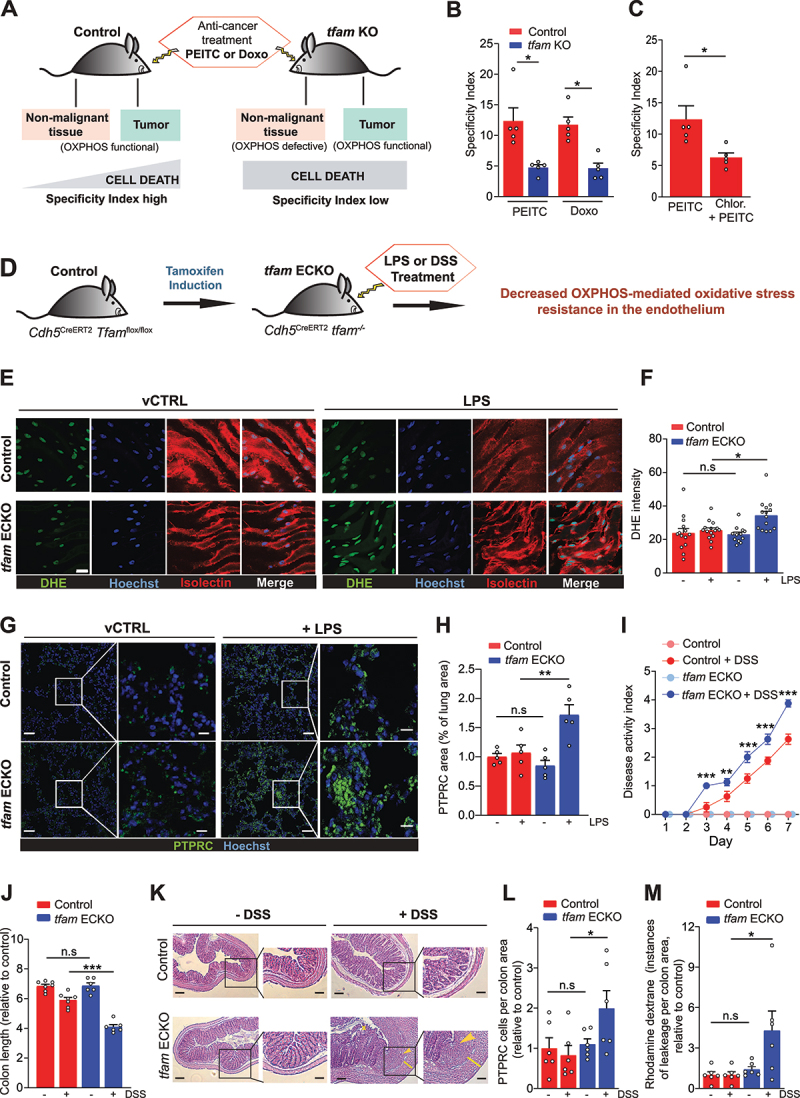
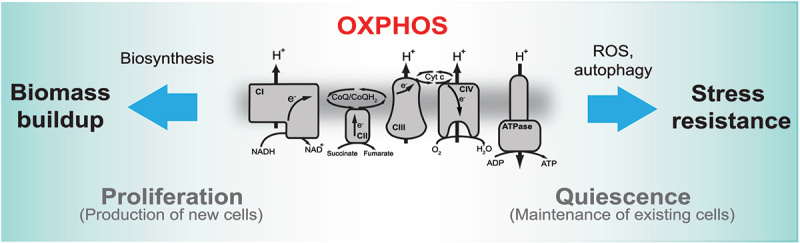
Mitochondrial oxidative phosphorylation (OXPHOS) generates ATP, but OXPHOS also supports biosynthesis during proliferation. In contrast, the role of OXPHOS during quiescence, beyond ATP production, is not well understood. Using mouse models of inducible OXPHOS deficiency in all cell types or specifically in the vascular endothelium that negligibly relies on OXPHOS-derived ATP, we show that selectively during quiescence OXPHOS provides oxidative stress resistance by supporting macroautophagy/autophagy. Mechanistically, OXPHOS constitutively generates low levels of endogenous ROS that induce autophagy via attenuation of ATG4B activity, which provides protection from ROS insult. Physiologically, the OXPHOS-autophagy system (i) protects healthy tissue from toxicity of ROS-based anticancer therapy, and (ii) provides ROS resistance in the endothelium, ameliorating systemic LPS-induced inflammation as well as inflammatory bowel disease. Hence, cells acquired mitochondria during evolution to profit from oxidative metabolism, but also built in an autophagy-based ROS-induced protective mechanism to guard against oxidative stress associated with OXPHOS function during quiescence.Abbreviations: AMPK: AMP-activated protein kinase; AOX: alternative oxidase; Baf A: bafilomycin A1; CI, respiratory complexes I; DCF-DA: 2',7'-dichlordihydrofluorescein diacetate; DHE: dihydroethidium; DSS: dextran sodium sulfate; ΔΨmi: mitochondrial inner membrane potential; EdU: 5-ethynyl-2'-deoxyuridine; ETC: electron transport chain; FA: formaldehyde; HUVEC; human umbilical cord endothelial cells; IBD: inflammatory bowel disease; LC3B: microtubule associated protein 1 light chain 3 beta; LPS: lipopolysaccharide; MEFs: mouse embryonic fibroblasts; MTORC1: mechanistic target of rapamycin kinase complex 1; mtDNA: mitochondrial DNA; NAC: N-acetyl cysteine; OXPHOS: oxidative phosphorylation; PCs: proliferating cells; PE: phosphatidylethanolamine; PEITC: phenethyl isothiocyanate; QCs: quiescent cells; ROS: reactive oxygen species; PLA2: phospholipase A2, WB: western blot.
Keywords: ATG4B; biosynthesis; cell death; electron transport chain; endothelial cells; mitochondria; oxidative phosphorylation; oxidative stress; reactive oxygen species.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
-
- Kops GJ, Dansen TB, Polderman PE. et al. Forkhead transcription factor FOXO3a protects quiescent cells from oxidative stress. Nature. 2002;419(6904):316–321. PubMed PMID: 12239572 - PubMed
-
- Naderi J, Hung M, Pandey S.. Oxidative stress-induced apoptosis in dividing fibroblasts involves activation of p38 MAP kinase and over-expression of Bax: resistance of quiescent cells to oxidative stress. Apoptosis. 2003;8(1):91–100. PubMed PMID: 12510156 - PubMed
-
- Valentin M, Yang E.. Autophagy is activated, but is not required for the G0 function of BCL-2 or BCL-xL. Cell Cycle. 2008;7(17):2762–2768. PubMed PMID: 18758240 - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials