

Genome-wide cis-decoding for expression design in tomato using cistrome data and explainable deep learning
- PMID: 35258588
- PMCID: PMC9134063
- DOI: 10.1093/plcell/koac079
Genome-wide cis-decoding for expression design in tomato using cistrome data and explainable deep learning
Abstract
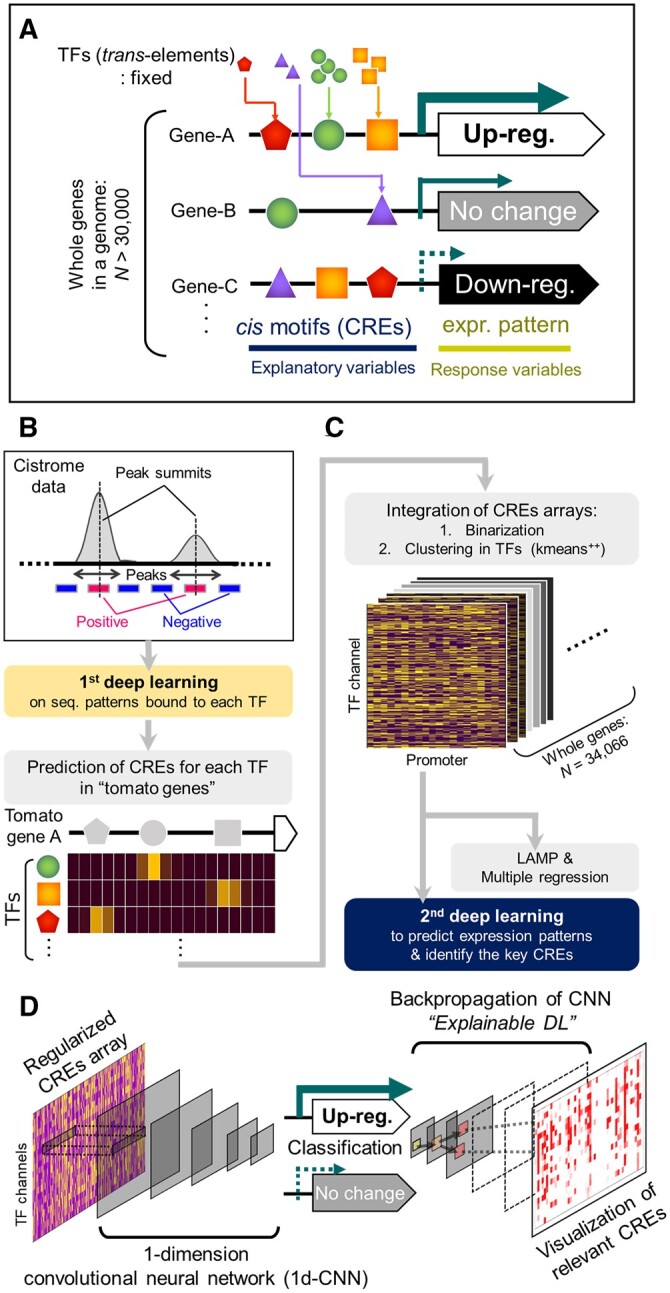
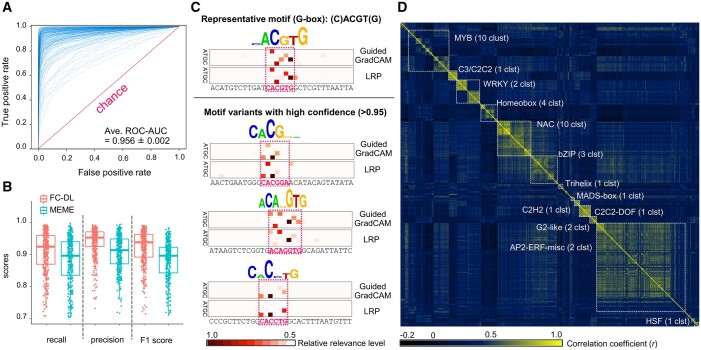
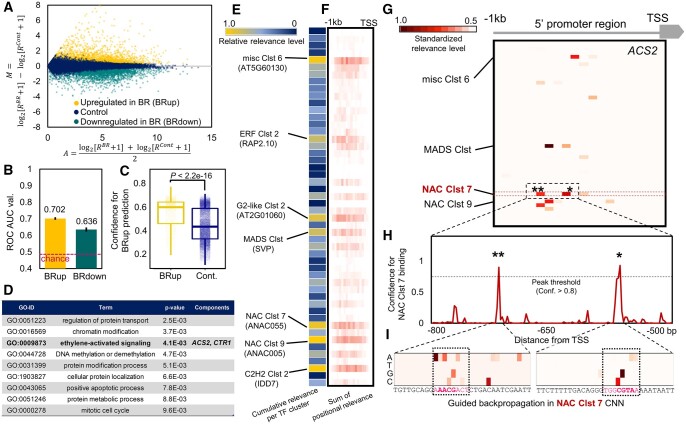
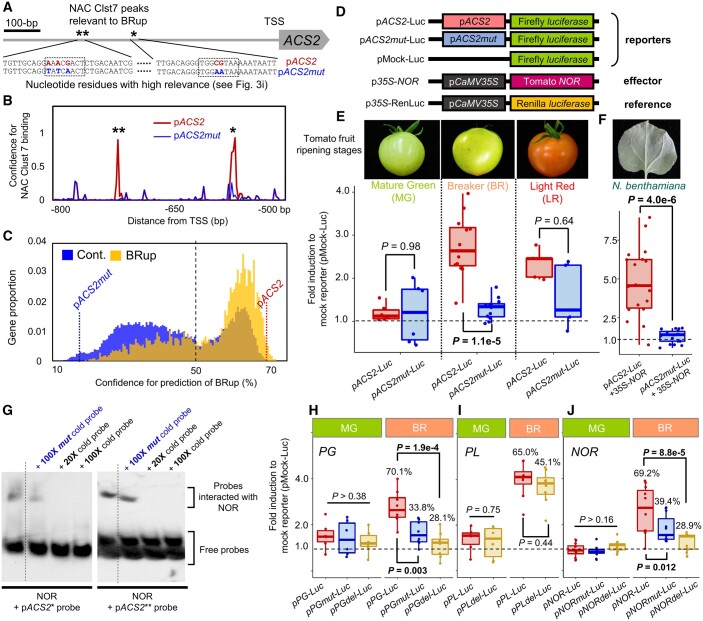
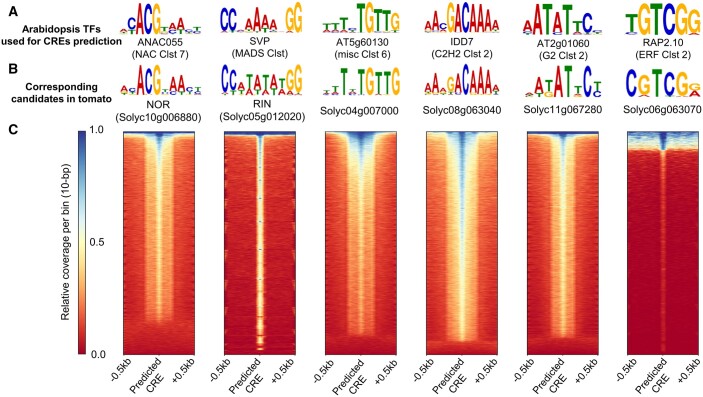
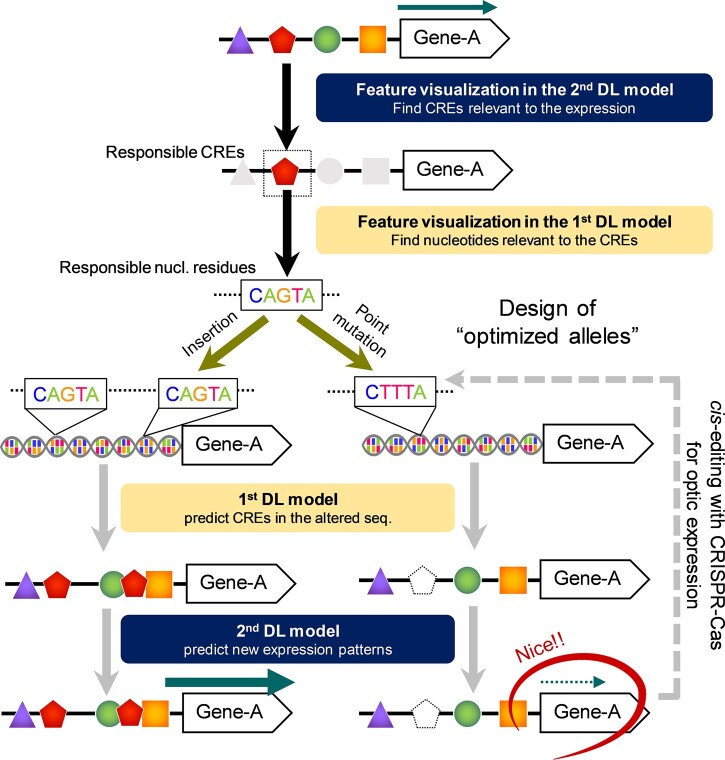
In the evolutionary history of plants, variation in cis-regulatory elements (CREs) resulting in diversification of gene expression has played a central role in driving the evolution of lineage-specific traits. However, it is difficult to predict expression behaviors from CRE patterns to properly harness them, mainly because the biological processes are complex. In this study, we used cistrome datasets and explainable convolutional neural network (CNN) frameworks to predict genome-wide expression patterns in tomato (Solanum lycopersicum) fruit from the DNA sequences in gene regulatory regions. By fixing the effects of trans-acting factors using single cell-type spatiotemporal transcriptome data for the response variables, we developed a prediction model for crucial expression patterns in the initiation of tomato fruit ripening. Feature visualization of the CNNs identified nucleotide residues critical to the objective expression pattern in each gene, and their effects were validated experimentally in ripening tomato fruit. This cis-decoding framework will not only contribute to the understanding of the regulatory networks derived from CREs and transcription factor interactions, but also provides a flexible means of designing alleles for optimized expression.
© The Author(s) 2022. Published by Oxford University Press on behalf of American Society of Plant Biologists.
Figures
Comment in
-
Decoding the cis-regulation of tomato fruit development with deep learning.Plant Cell. 2022 May 24;34(6):2108-2109. doi: 10.1093/plcell/koac094. Plant Cell. 2022. PMID: 35325244 Free PMC article. No abstract available.
References
-
- Akagi T, Onishi M, Masuda K, Kuroki R, Baba K, Takeshita K, Suzuki T, Niikawa T, Uchida S, Ise T (2020) Explainable deep learning reproduces a ‘professional eye’ on the diagnosis of internal disorders in persimmon fruit. Plant Cell Physiol 61: 1967–1973 - PubMed
-
- Alber M, Lapuschkin S, Seegerer P, Hägele M, Schütt KT, Montavon G, Samek W, Müller KR, Dähne S, Kindermans PJ (2019) iNNvestigate neural networks! J Mach Learn Res 20: 1–8
-
- Alipanahi B, Delong A, Weirauch MT, Frey BJ (2015) Predicting the sequence specificities of DNA-and RNA-binding proteins by deep learning. Nat Biotechnol 33: 831–838 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
