

Genetic characteristics of 234 Italian patients with macular and cone/cone-rod dystrophy
- PMID: 35260635
- PMCID: PMC8904500
- DOI: 10.1038/s41598-022-07618-1
Genetic characteristics of 234 Italian patients with macular and cone/cone-rod dystrophy
Abstract
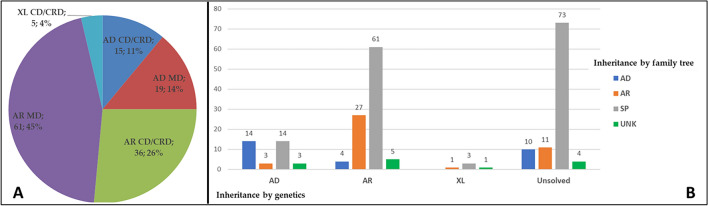
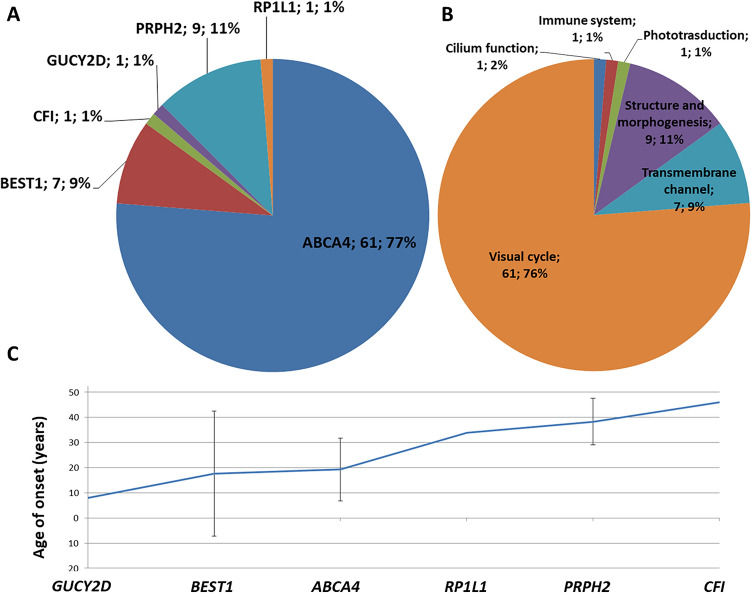
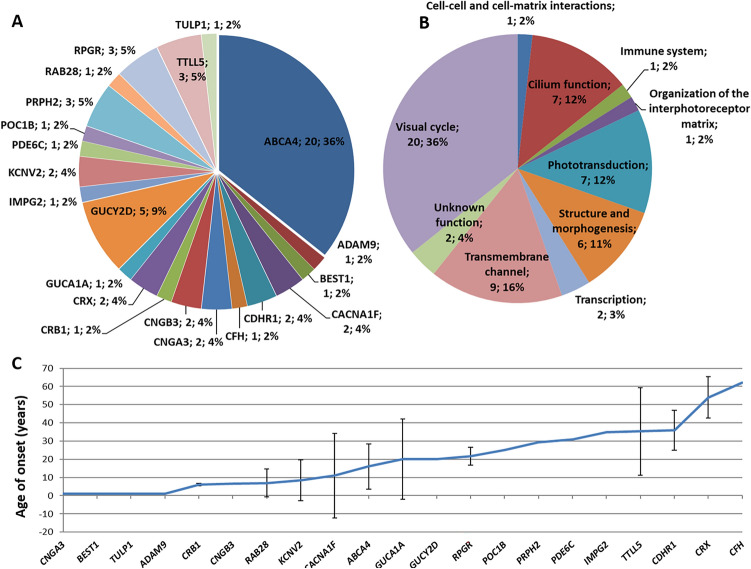
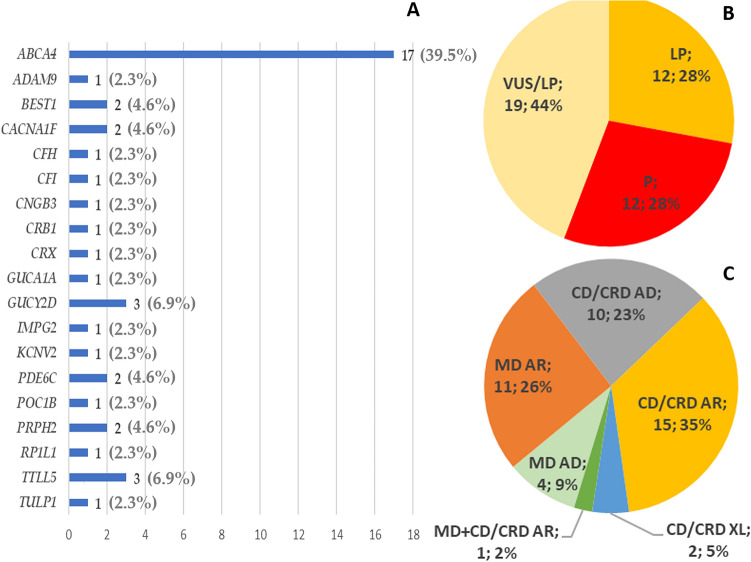
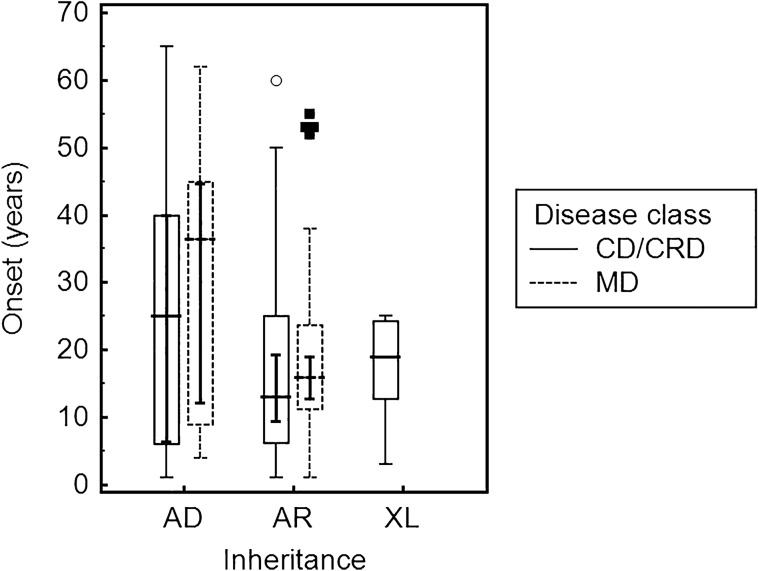
Two-hundred and thirty-four Italian patients with a clinical diagnosis of macular, cone and cone-rod dystrophies (MD, CD, and CRD) were examined using next-generation sequencing (NGS) and gene sequencing panels targeting a specific set of genes, Sanger sequencing and-when necessary-multiplex ligation-dependent probe amplification (MLPA) to diagnose the molecular cause of the aforementioned diseases. When possible, segregation analysis was performed in order to confirm unsolved cases. Each patient's retinal phenotypic characteristics were determined using focal and full-field ERGs, perimetry, spectral domain optical coherence tomography and fundus autofluorescence. We identified 236 potentially causative variants in 136 patients representing the 58.1% of the total cohort, 43 of which were unpublished. After stratifying the patients according to their clinical suspicion, the diagnostic yield was 62.5% and 53.8% for patients with MD and for those with CD/CRD, respectively. The mode of inheritance of all cases confirmed by genetic analysis was 70% autosomal recessive, 26% dominant, and 4% X-linked. The main cause (59%) of both MD and CD/CRD cases was the presence of variants in the ABCA4 gene, followed by variants in PRPH2 (9%) and BEST1 (6%). A careful morpho-functional evaluation of the phenotype, together with genetic counselling, resulted in an acceptable diagnostic yield in a large cohort of Italian patients. Our study emphasizes the role of targeted NGS to diagnose MDs, CDs, and CRDs, as well as the clinical usefulness of segregation analysis for patients with unsolved diagnosis.
© 2022. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
Similar articles
-
Novel variants identified with next-generation sequencing in Polish patients with cone-rod dystrophy.Mol Vis. 2018 Apr 26;24:326-339. eCollection 2018. Mol Vis. 2018. PMID: 29769798 Free PMC article.
-
Characterization of the cone-rod dystrophy retinal phenotype caused by novel homozygous DRAM2 mutations.Exp Eye Res. 2019 Oct;187:107752. doi: 10.1016/j.exer.2019.107752. Epub 2019 Aug 5. Exp Eye Res. 2019. PMID: 31394102
-
SPATA7: Evolving phenotype from cone-rod dystrophy to retinitis pigmentosa.Ophthalmic Genet. 2016 Sep;37(3):333-8. doi: 10.3109/13816810.2015.1130154. Epub 2016 Feb 8. Ophthalmic Genet. 2016. PMID: 26854980 Free PMC article.
-
[From gene to disease: from the ABCA4 gene to Stargardt disease, cone-rod dystrophy and retinitis pigmentosa].Ned Tijdschr Geneeskd. 2002 Aug 24;146(34):1581-4. Ned Tijdschr Geneeskd. 2002. PMID: 12224481 Review. Dutch.
-
Cone rod dystrophies.Orphanet J Rare Dis. 2007 Feb 1;2:7. doi: 10.1186/1750-1172-2-7. Orphanet J Rare Dis. 2007. PMID: 17270046 Free PMC article. Review.
Cited by
-
A novel homozygous splice site variant in ARL2BP causes a syndromic autosomal recessive rod-cone dystrophy with situs inversus, asthenozoospermia, unilateral renal agenesis and microcysts.BMC Med Genomics. 2024 Apr 22;17(1):100. doi: 10.1186/s12920-024-01868-w. BMC Med Genomics. 2024. PMID: 38649918 Free PMC article.
-
Multimodal Study of PRPH2 Gene-Related Retinal Phenotypes.Diagnostics (Basel). 2022 Jul 31;12(8):1851. doi: 10.3390/diagnostics12081851. Diagnostics (Basel). 2022. PMID: 36010202 Free PMC article.
-
Identification of a novel large multigene deletion and a frameshift indel in PDE6B as the underlying cause of early-onset recessive rod-cone degeneration.Cold Spring Harb Mol Case Stud. 2022 Dec 28;8(7):a006247. doi: 10.1101/mcs.a006247. Print 2022 Dec. Cold Spring Harb Mol Case Stud. 2022. PMID: 36376065 Free PMC article.
-
Stargardt Disease Due to an Intronic Mutation in the ABCA4: A Case Report.Int Med Case Rep J. 2022 Nov 29;15:693-698. doi: 10.2147/IMCRJ.S391001. eCollection 2022. Int Med Case Rep J. 2022. PMID: 36471740 Free PMC article.
-
Genetics of Inherited Retinal Diseases in Understudied Ethnic Groups in Italian Hospitals.Front Genet. 2022 Jun 28;13:914345. doi: 10.3389/fgene.2022.914345. eCollection 2022. Front Genet. 2022. PMID: 35836572 Free PMC article.
References
-
- Talib M, et al. Clinical and genetic characteristics of male patients with RPGR-associated retinal dystrophies: A long-term follow-up study. Retina. 2019;39:1186–1199. - PubMed
-
- Cremers FP, et al. Autosomal recessive retinitis pigmentosa and cone-rod dystrophy caused by splice site mutations in the Stargardt’s disease gene ABCR. Hum. Mol. Genet. 1998;7:355–362. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
