

Sestrin2 protects against cholestatic liver injury by inhibiting endoplasmic reticulum stress and NLRP3 inflammasome-mediated pyroptosis
- PMID: 35260799
- PMCID: PMC8980001
- DOI: 10.1038/s12276-022-00737-9
Sestrin2 protects against cholestatic liver injury by inhibiting endoplasmic reticulum stress and NLRP3 inflammasome-mediated pyroptosis
Abstract
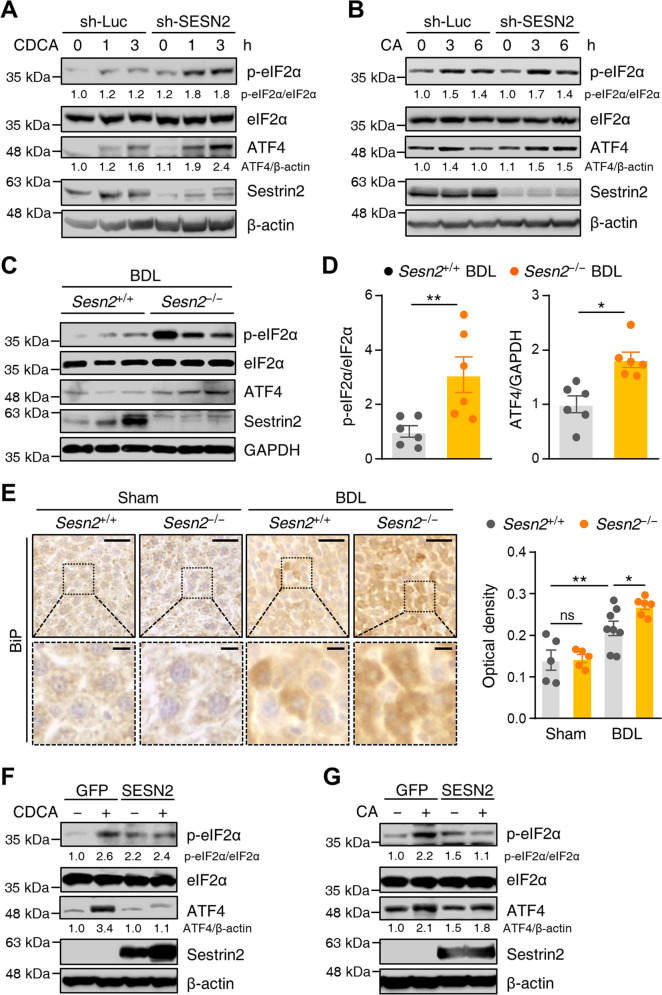
Chronic exposure to bile acid in the liver due to impaired bile flow induces cholestatic liver disease, resulting in hepatotoxicity and liver fibrosis. Sestrin2, a highly conserved, stress-inducible protein, has been implicated in cellular responses to multiple stress conditions and the maintenance of cellular homeostasis. However, its role in cholestatic liver injury is not fully understood. In this study, we investigated the role of hepatic Sestrin2 in cholestatic liver injury and its underlying mechanisms using in vivo and in vitro approaches. Hepatic Sestrin2 expression was upregulated by activating transcription factor 4 (ATF4) and CCAAT/enhancer-binding protein-β (C/EBP-β) after treatment with bile acids and correlated with endoplasmic reticulum (ER) stress responses. Bile-duct ligation (BDL)-induced hepatocellular apoptosis and liver fibrosis were exacerbated in Sestrin2-knockout (Sesn2-/-) mice. Moreover, Sestrin2 deficiency enhanced cholestasis-induced hepatic ER stress, whereas Sestrin2 overexpression ameliorated bile acid-induced ER stress. Notably, the mammalian target of rapamycin (mTOR) inhibitor rapamycin and the AMP-activated protein kinase (AMPK) activator AICAR reversed bile acid-induced ER stress in Sestrin2-deficient cells. Furthermore, Sestrin2 deficiency promoted cholestasis-induced hepatic pyroptosis by activating NLRP3 inflammasomes. Thus, our study provides evidence for the biological significance of Sestrin2 and its relationship with cholestatic liver injury, suggesting the potential role of Sestrin2 in regulating ER stress and inflammasome activation during cholestatic liver injury.
© 2022. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures







References
-
- Haenel LC, Bradway WR, Costantini PJ. Thrombophlebitis complicating sternocostoclavicular hyperostosis. Postgrad. Med. 1980;68:113–115. - PubMed
-
- Zollner G, Trauner M. Mechanisms of cholestasis. Clin. Liver Dis. 2008;12:1–26. - PubMed
-
- Kim S, et al. Impaired autophagy promotes bile acid-induced hepatic injury and accumulation of ubiquitinated proteins. Biochem. Biophys. Res. Commun. 2018;495:1541–1547. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
