

The 2019 and 2021 International Workshops on Alport Syndrome
- PMID: 35260866
- PMCID: PMC8904161
- DOI: 10.1038/s41431-022-01075-0
The 2019 and 2021 International Workshops on Alport Syndrome
Erratum in
-
Correction: The 2019 and 2021 International workshops on Alport syndrome.Eur J Hum Genet. 2024 Jan;32(1):130. doi: 10.1038/s41431-023-01286-z. Eur J Hum Genet. 2024. PMID: 36690832 Free PMC article. No abstract available.
Abstract
In 1927 Arthur Cecil Alport, a South African physician, described a British family with an inherited form of kidney disease that affected males more severely than females and was sometimes associated with hearing loss. In 1961, the eponymous name Alport syndrome was adopted. In the late twentieth century three genes responsible for the disease were discovered: COL4A3, COL4A4, and COL4A5 encoding for the α3, α4, α5 polypeptide chains of type IV collagen, respectively. These chains assemble to form heterotrimers of type IV collagen in the glomerular basement membrane. Scientists, clinicians, patient representatives and their families, and pharma companies attended the 2019 International Workshop on Alport Syndrome, held in Siena, Italy, from October 22 to 26, and the 2021 online Workshop from November 30 to December 4. The main topics included: disease re-naming, acknowledging the need to identify an appropriate term able to reflect considerable clinical variability; a strategy for increasing the molecular diagnostic rate; genotype-phenotype correlation from monogenic to digenic forms; new therapeutics and new therapeutic approaches; and gene therapy using gene editing. The exceptional collaborative climate that was established in the magical medieval setting of Siena continued in the online workshop of 2021. Conditions were established for collaborations between leading experts in the sector, including patients and drug companies, with the aim of identifying a cure for Alport syndrome.
Conflict of interest statement
The authors declare no competing interests.
Figures
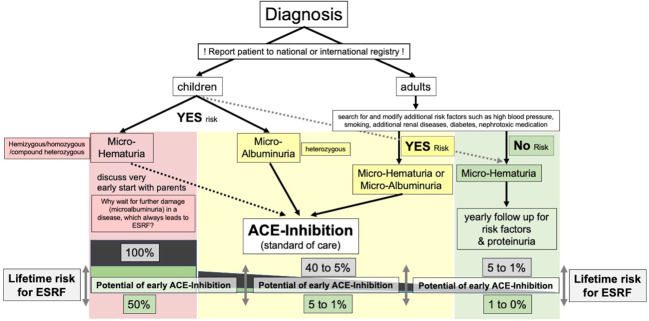
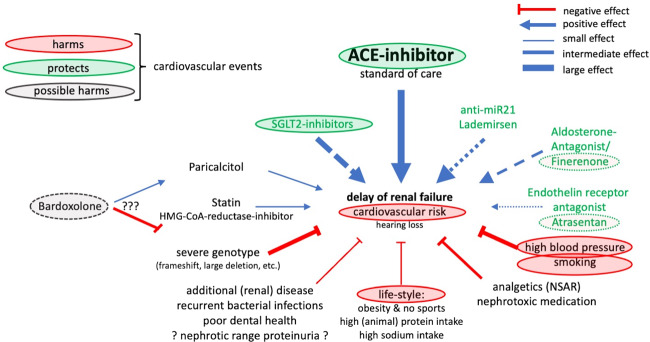

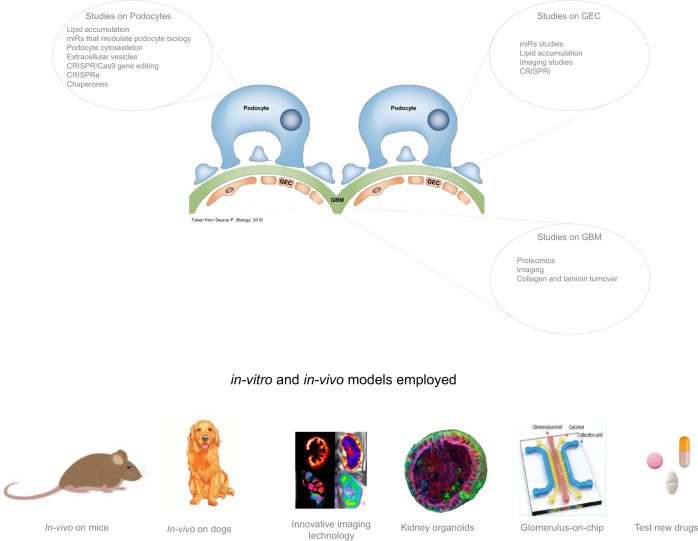
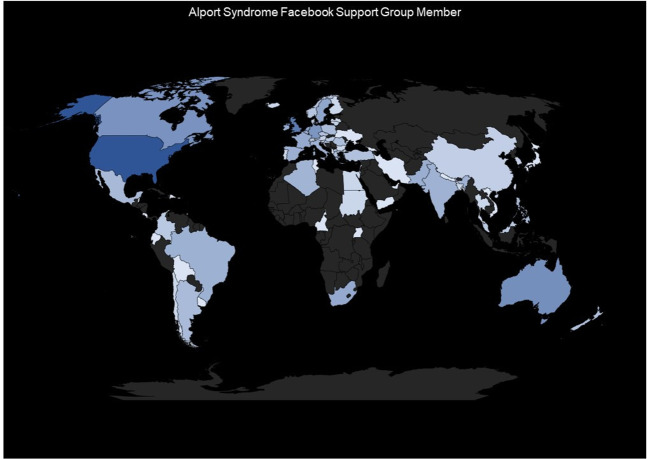
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
