

Rapid expansion and extinction of antibiotic resistance mutations during treatment of acute bacterial respiratory infections
- PMID: 35264582
- PMCID: PMC8907320
- DOI: 10.1038/s41467-022-28188-w
Rapid expansion and extinction of antibiotic resistance mutations during treatment of acute bacterial respiratory infections
Abstract
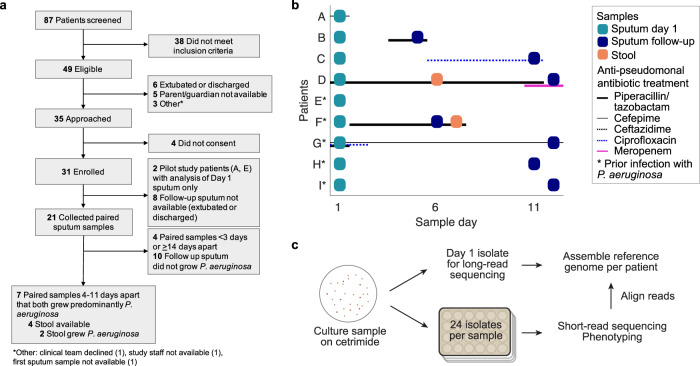
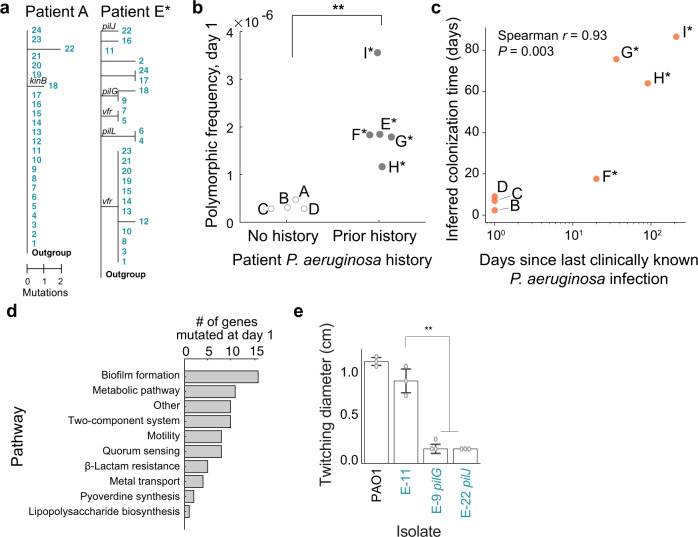
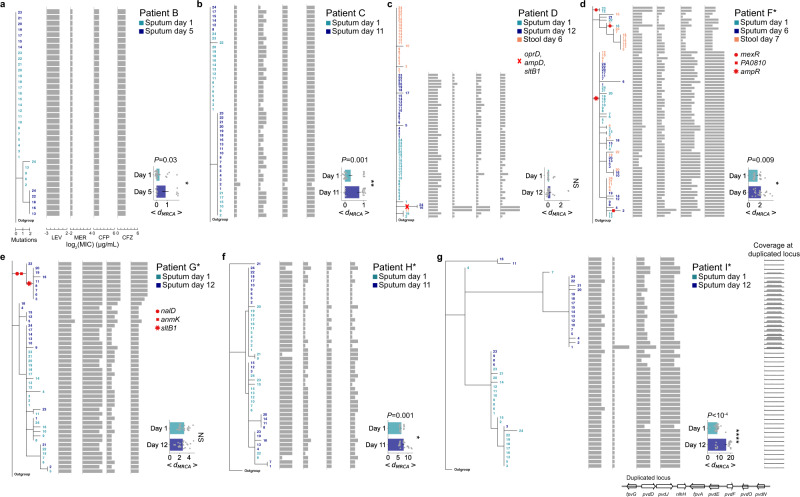
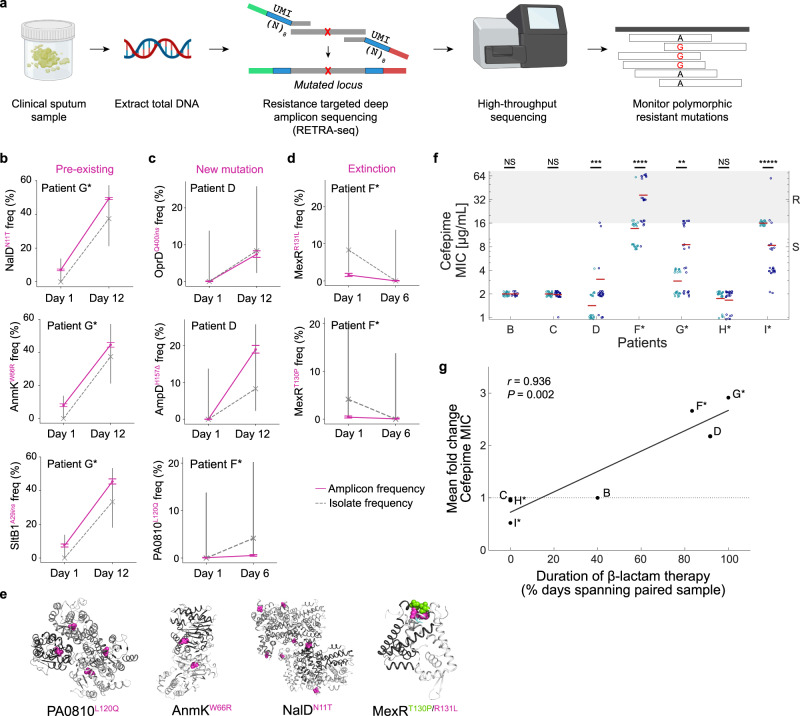
Acute bacterial infections are often treated empirically, with the choice of antibiotic therapy updated during treatment. The effects of such rapid antibiotic switching on the evolution of antibiotic resistance in individual patients are poorly understood. Here we find that low-frequency antibiotic resistance mutations emerge, contract, and even go to extinction within days of changes in therapy. We analyzed Pseudomonas aeruginosa populations in sputum samples collected serially from 7 mechanically ventilated patients at the onset of respiratory infection. Combining short- and long-read sequencing and resistance phenotyping of 420 isolates revealed that while new infections are near-clonal, reflecting a recent colonization bottleneck, resistance mutations could emerge at low frequencies within days of therapy. We then measured the in vivo frequencies of select resistance mutations in intact sputum samples with resistance-targeted deep amplicon sequencing (RETRA-Seq), which revealed that rare resistance mutations not detected by clinically used culture-based methods can increase by nearly 40-fold over 5-12 days in response to antibiotic changes. Conversely, mutations conferring resistance to antibiotics not administered diminish and even go to extinction. Our results underscore how therapy choice shapes the dynamics of low-frequency resistance mutations at short time scales, and the findings provide a possibility for driving resistance mutations to extinction during early stages of infection by designing patient-specific antibiotic cycling strategies informed by deep genomic surveillance.
© 2022. The Author(s).
Conflict of interest statement
H.C., M.M.S., R.K., and G.P.P. are co-inventors on a provisional patent application filed on RETRA-Seq.
Figures
References
-
- Levy SB, Marshall B. Antibacterial resistance worldwide: causes, challenges and responses. Nat. Med. 2004;10:S122–S129. - PubMed
-
- Marvig RL, Sommer LM, Molin S, Johansen HK. Convergent evolution and adaptation of Pseudomonas aeruginosa within patients with cystic fibrosis. Nat. Genet. 2015;47:57–64. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
