

Therapy resistance: opportunities created by adaptive responses to targeted therapies in cancer
- PMID: 35264777
- PMCID: PMC9149051
- DOI: 10.1038/s41568-022-00454-5
Therapy resistance: opportunities created by adaptive responses to targeted therapies in cancer
Abstract
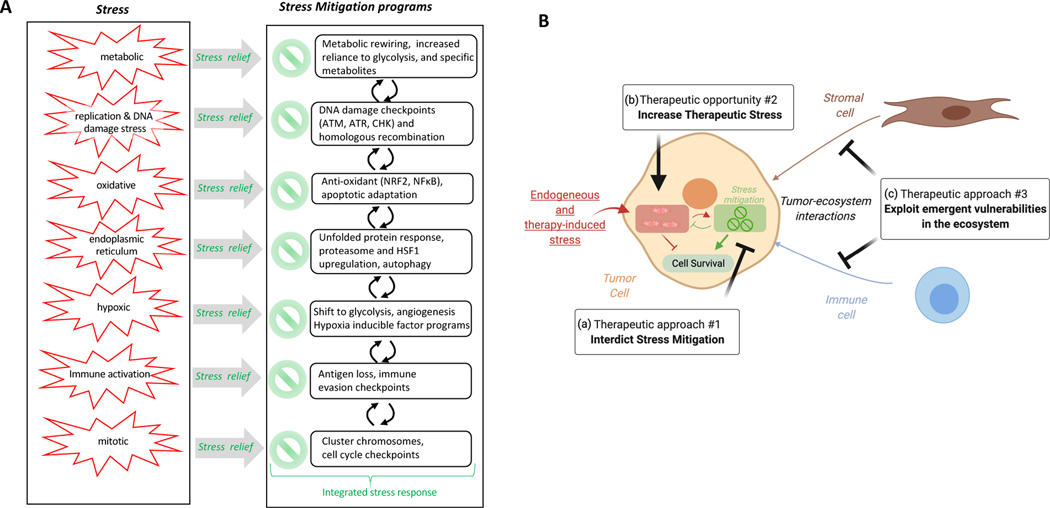
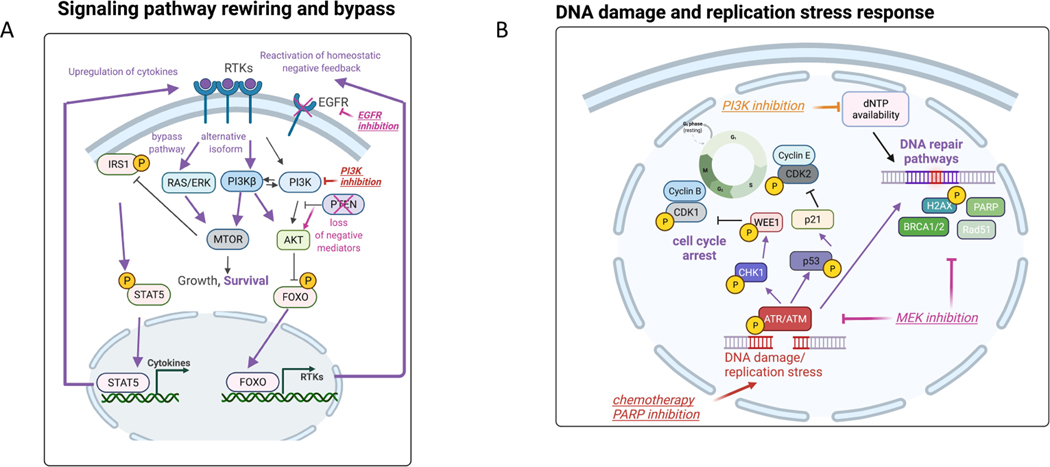
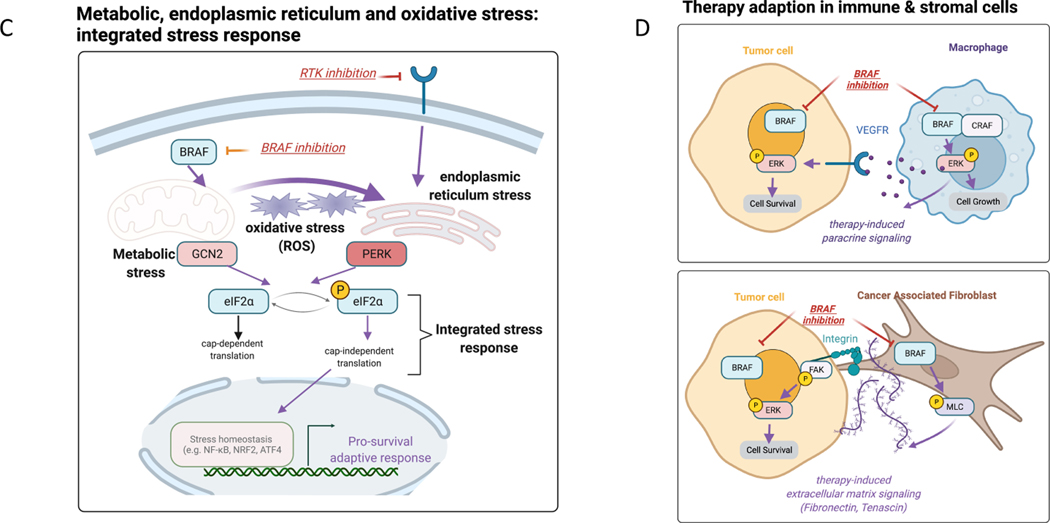
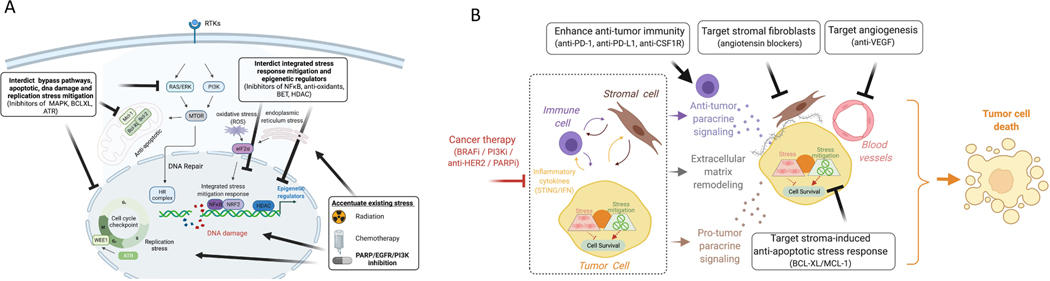
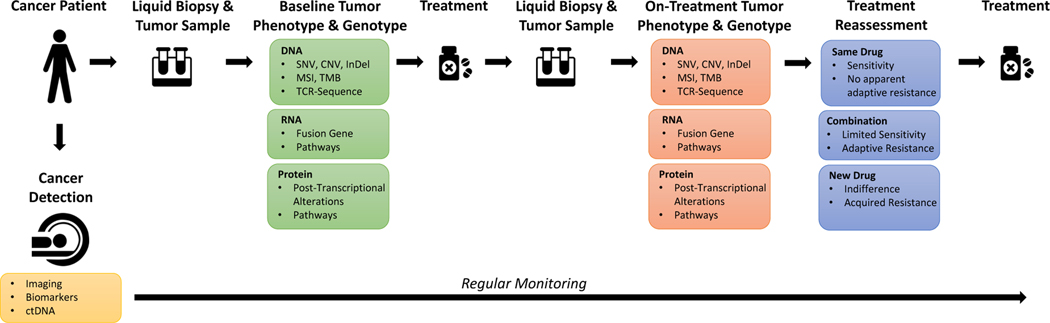
Normal cells explore multiple states to survive stresses encountered during development and self-renewal as well as environmental stresses such as starvation, DNA damage, toxins or infection. Cancer cells co-opt normal stress mitigation pathways to survive stresses that accompany tumour initiation, progression, metastasis and immune evasion. Cancer therapies accentuate cancer cell stresses and invoke rapid non-genomic stress mitigation processes that maintain cell viability and thus represent key targetable resistance mechanisms. In this Review, we describe mechanisms by which tumour ecosystems, including cancer cells, immune cells and stroma, adapt to therapeutic stresses and describe three different approaches to exploit stress mitigation processes: (1) interdict stress mitigation to induce cell death; (2) increase stress to induce cellular catastrophe; and (3) exploit emergent vulnerabilities in cancer cells and cells of the tumour microenvironment. We review challenges associated with tumour heterogeneity, prioritizing actionable adaptive responses for optimal therapeutic outcomes, and development of an integrative framework to identify and target vulnerabilities that arise from adaptive responses and engagement of stress mitigation pathways. Finally, we discuss the need to monitor adaptive responses across multiple scales and translation of combination therapies designed to take advantage of adaptive responses and stress mitigation pathways to the clinic.
© 2022. Springer Nature Limited.
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
