

Astrocytic junctional adhesion molecule-A regulates T-cell entry past the glia limitans to promote central nervous system autoimmune attack
- PMID: 35265839
- PMCID: PMC8899531
- DOI: 10.1093/braincomms/fcac044
Astrocytic junctional adhesion molecule-A regulates T-cell entry past the glia limitans to promote central nervous system autoimmune attack
Abstract
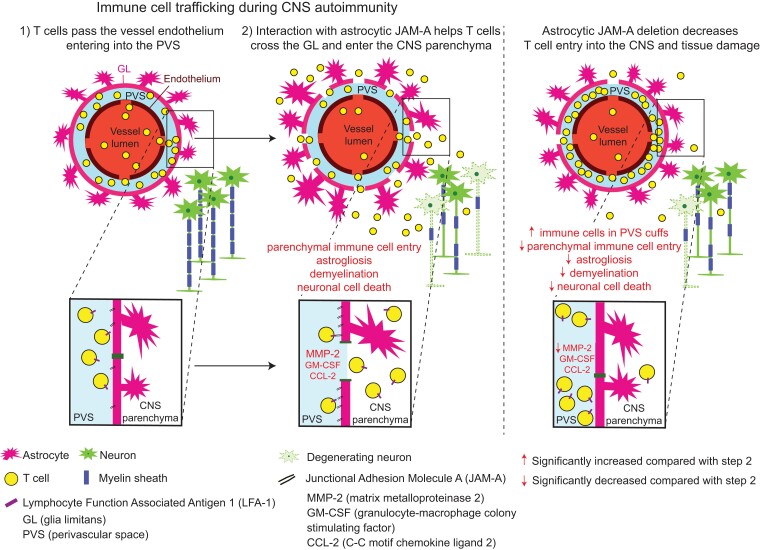
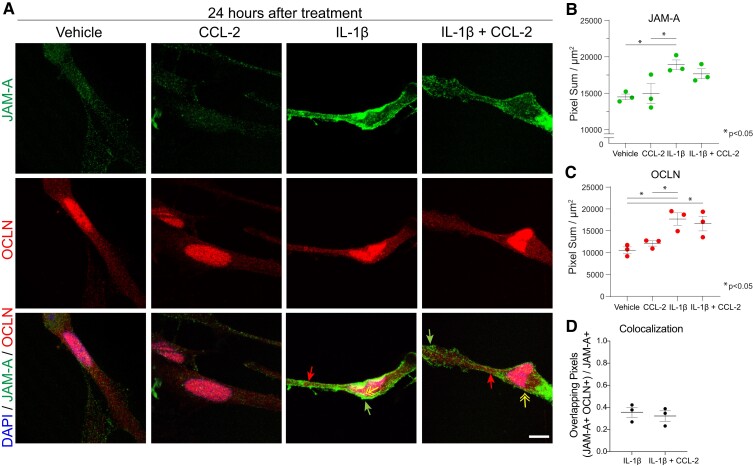
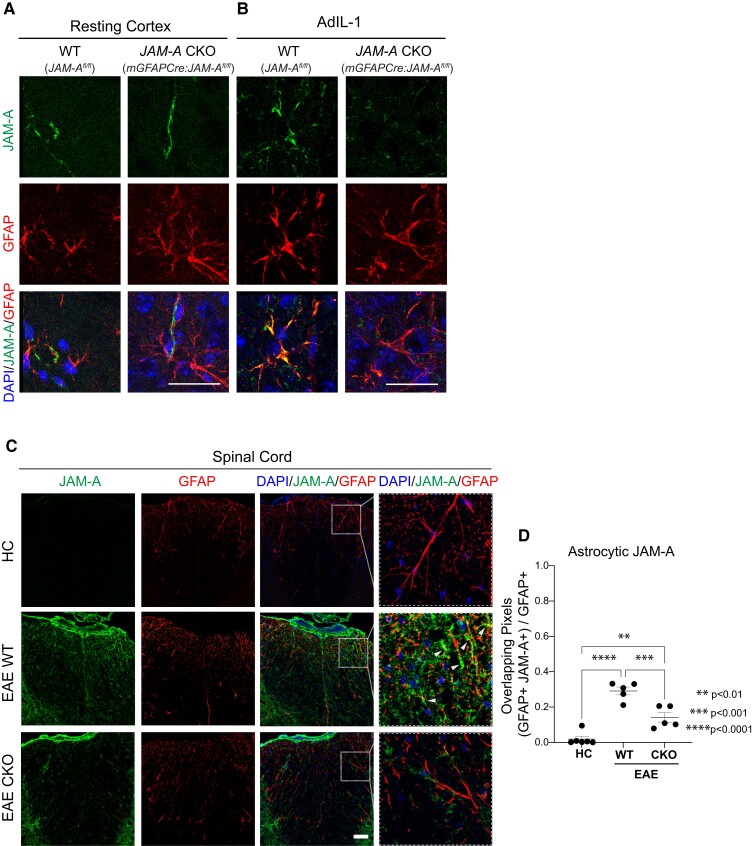
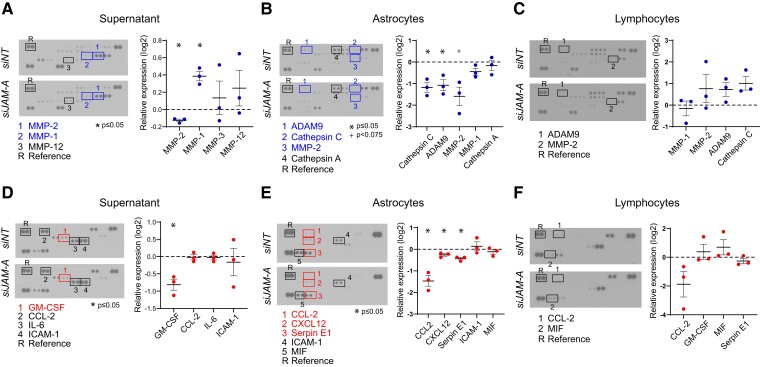
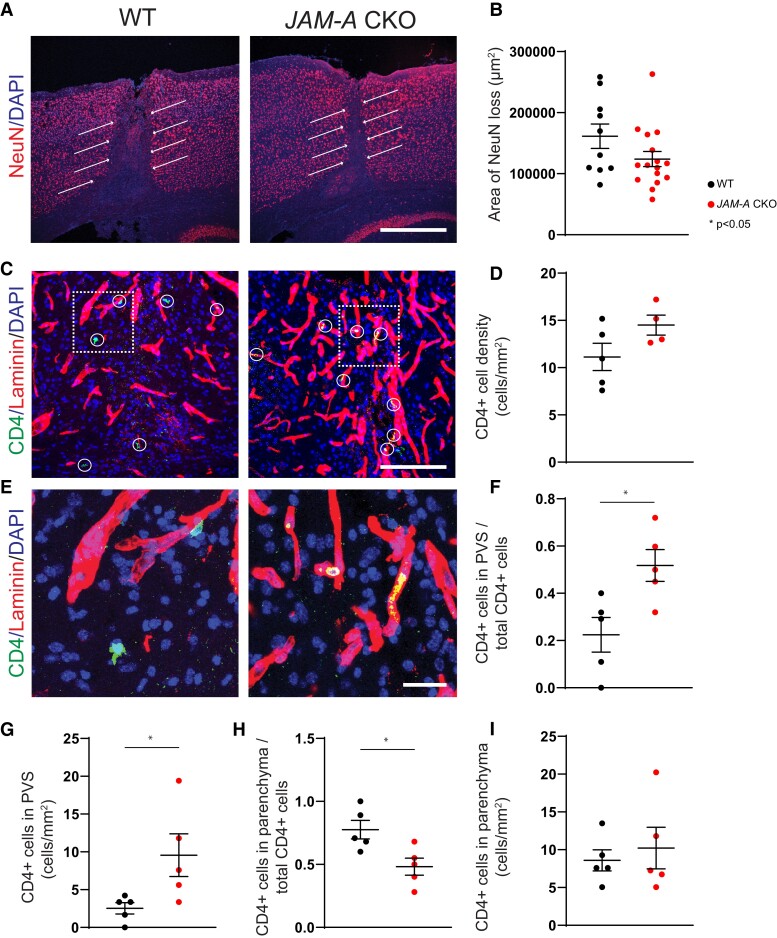
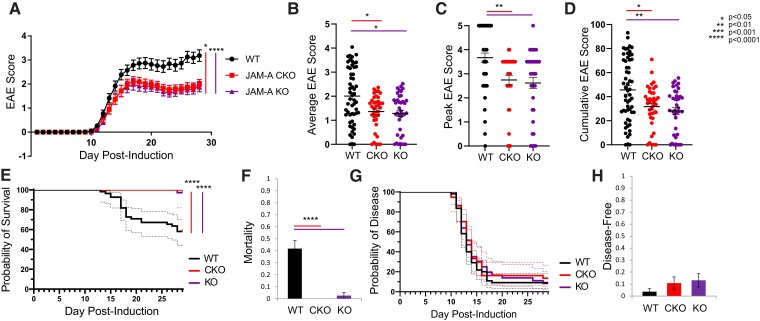
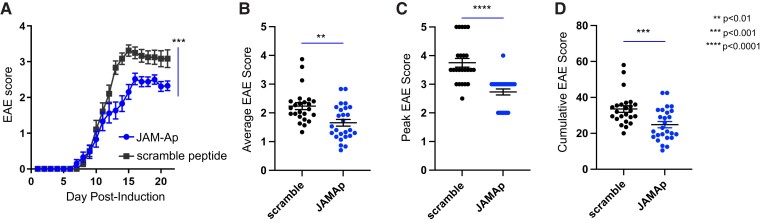
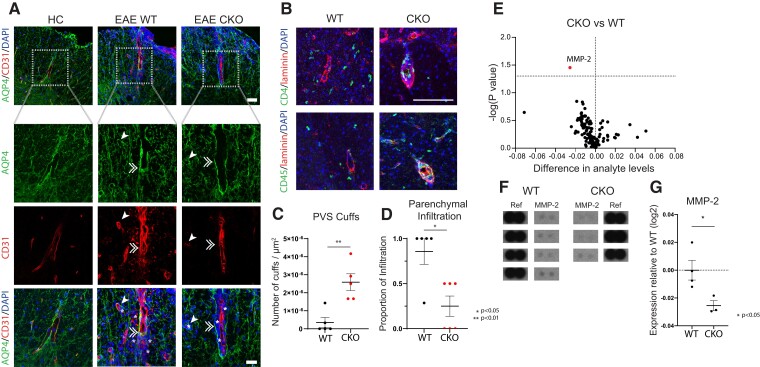
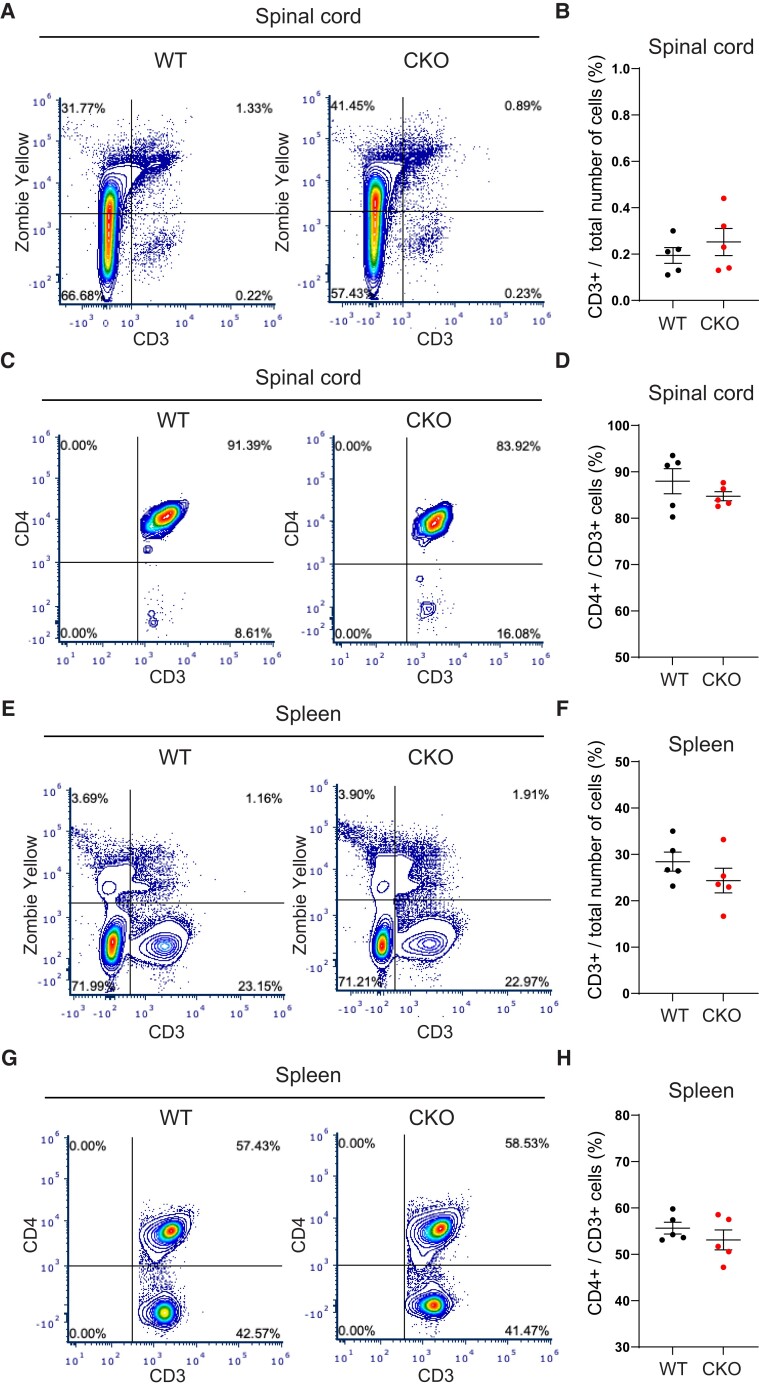
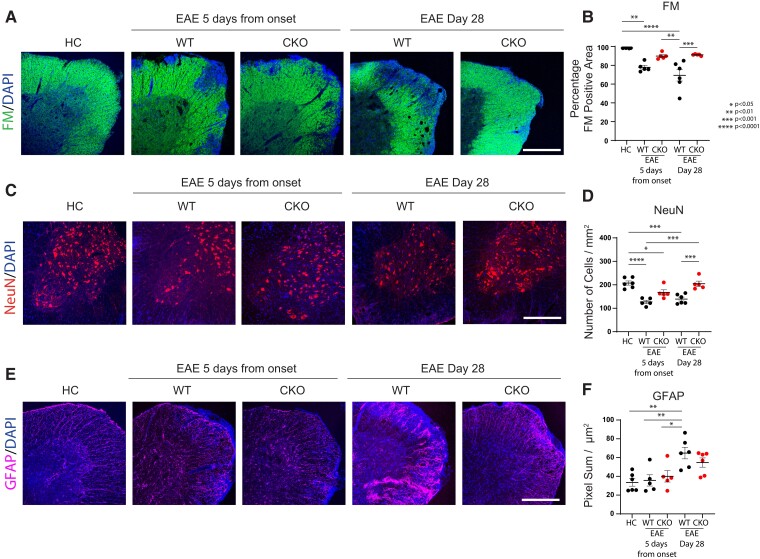
Contact-mediated interactions between the astrocytic endfeet and infiltrating immune cells within the perivascular space are underexplored, yet represent potential regulatory check-points against CNS autoimmune disease and disability. Reactive astrocytes upregulate junctional adhesion molecule-A, an immunoglobulin-like cell surface receptor that binds to T cells via its ligand, the integrin, lymphocyte function-associated antigen-1. Here, we tested the role of astrocytic junctional adhesion molecule-A in regulating CNS autoinflammatory disease. In cell co-cultures, we found that junctional adhesion molecule-A-mediated signalling between astrocytes and T cells increases levels of matrix metalloproteinase-2, C-C motif chemokine ligand 2 and granulocyte-macrophage colony-stimulating factor, pro-inflammatory factors driving lymphocyte entry and pathogenicity in multiple sclerosis and experimental autoimmune encephalomyelitis, an animal model of CNS autoimmune disease. In experimental autoimmune encephalomyelitis, mice with astrocyte-specific JAM-A deletion (mGFAP:CreJAM-Afl/fl ) exhibit decreased levels of matrix metalloproteinase-2, reduced ability of T cells to infiltrate the CNS parenchyma from the perivascular spaces and a milder histopathological and clinical course of disease compared with wild-type controls (JAM-Afl/fl ). Treatment of wild-type mice with intraperitoneal injection of soluble junctional adhesion molecule-A blocking peptide decreases the severity of experimental autoimmune encephalomyelitis, highlighting the potential of contact-mediated astrocyte-immune cell signalling as a novel translational target against neuroinflammatory disease.
Keywords: astrocyte immune cell cross-talk; experimental autoimmune encephalomyelitis; glia limitans; junctional adhesion molecule-A; multiple sclerosis.
© The Author(s) 2022. Published by Oxford University Press on behalf of the Guarantors of Brain.
Figures
References
-
- Abbott NJ, Rönnbäck L, Hansson E. Astrocyte–endothelial interactions at the blood–brain barrier. Nat Rev Neurosci. 2006;7(1):41–53. - PubMed
-
- Engelhardt B, Ransohoff RM. Capture, crawl, cross: The T cell code to breach the blood–brain barriers. Trends Immunol. 2012;33(12):579–589. - PubMed
-
- Schläger C, Körner H, Krueger M, et al. Effector T-cell trafficking between the leptomeninges and the cerebrospinal fluid. Nature. 2016;530(7590):349–353. - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous