

The Pathogenesis of Cardiac Fibrosis: A Review of Recent Progress
- PMID: 35269759
- PMCID: PMC8910720
- DOI: 10.3390/ijms23052617
The Pathogenesis of Cardiac Fibrosis: A Review of Recent Progress
Abstract
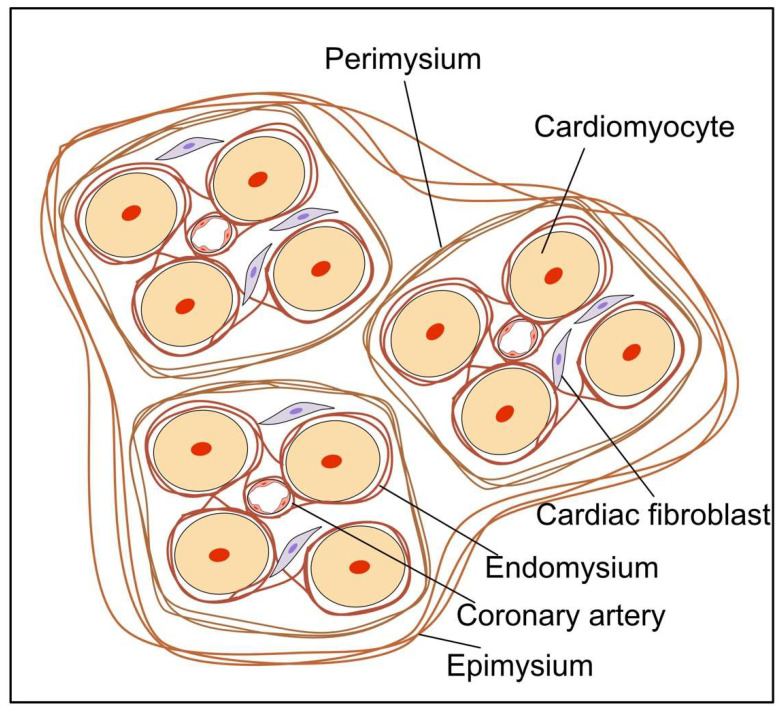
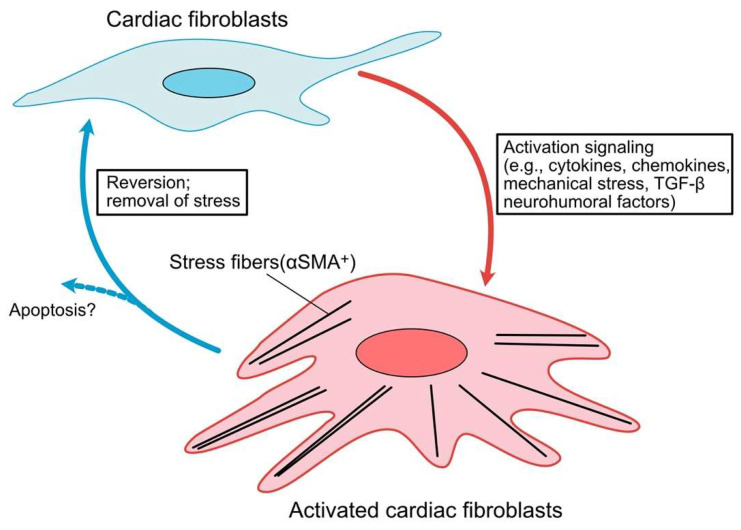
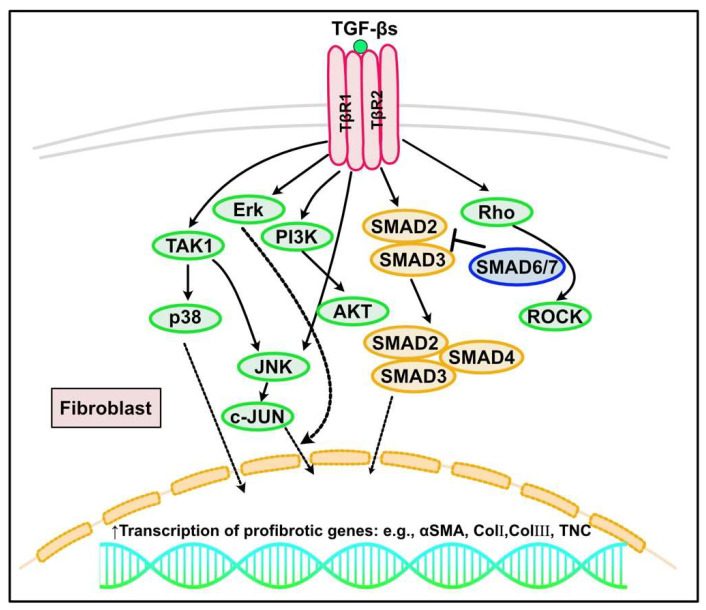
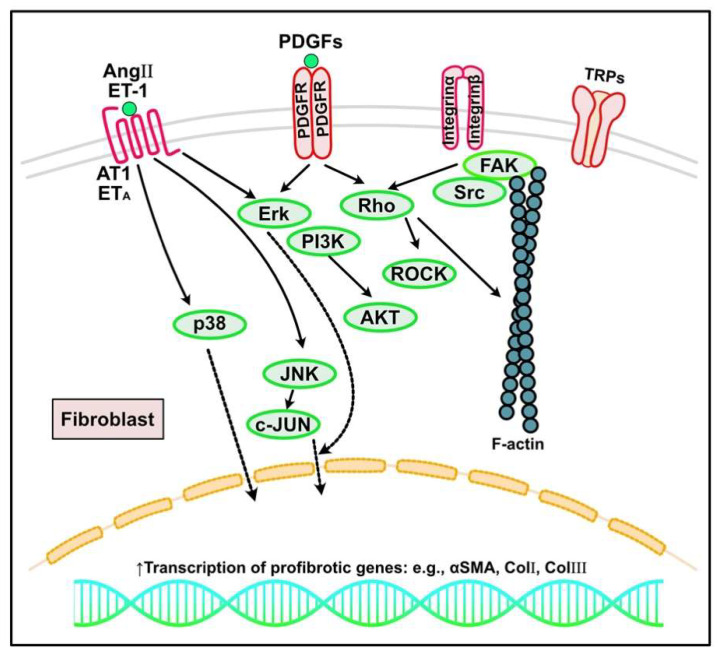
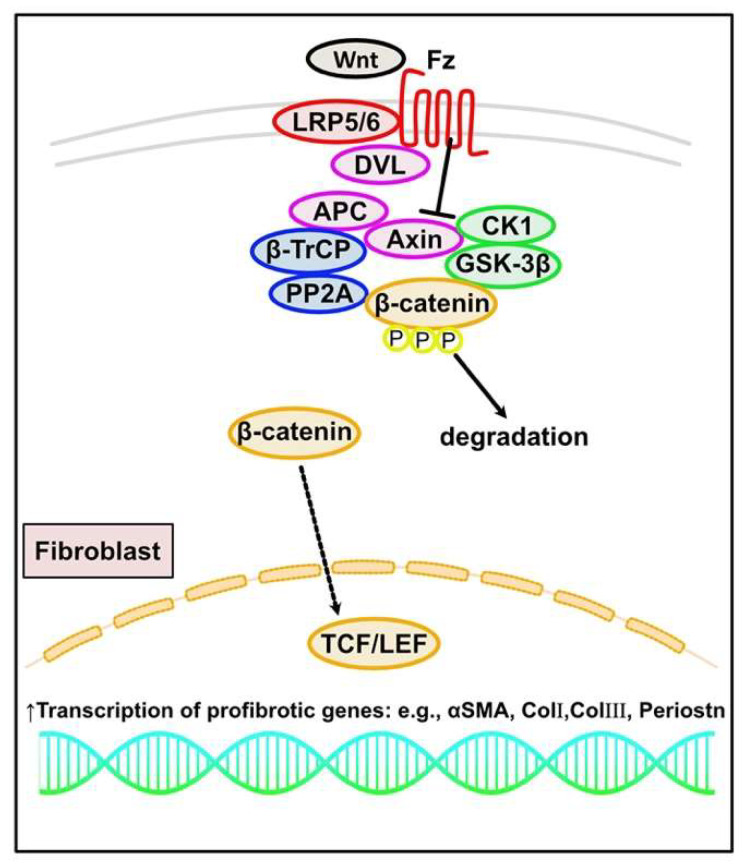
Fibrosis is defined as the excessive deposition of extracellular matrix (ECM) proteins in the interstitium. It is an essential pathological response to chronic inflammation. ECM protein deposition is initially protective and is critical for wound healing and tissue regeneration. However, pathological cardiac remodeling in excessive and continuous tissue damage with subsequent ECM deposition results in a distorted organ architecture and significantly impacts cardiac function. In this review, we summarized and discussed the histologic features of cardiac fibrosis with the signaling factors that control it. We evaluated the origin and characteristic markers of cardiac fibroblasts. We also discussed lymphatic vessels, which have become more important in recent years to improve cardiac fibrosis.
Keywords: cardiac fibrosis; marker of cardiac fibroblast; pathogenesis of cardiac fibrosis; signaling of cardiac fibrosis.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
