

Heterogeneity and clonality of kidney-infiltrating T cells in murine lupus nephritis
- PMID: 35271505
- PMCID: PMC9089785
- DOI: 10.1172/jci.insight.156048
Heterogeneity and clonality of kidney-infiltrating T cells in murine lupus nephritis
Abstract
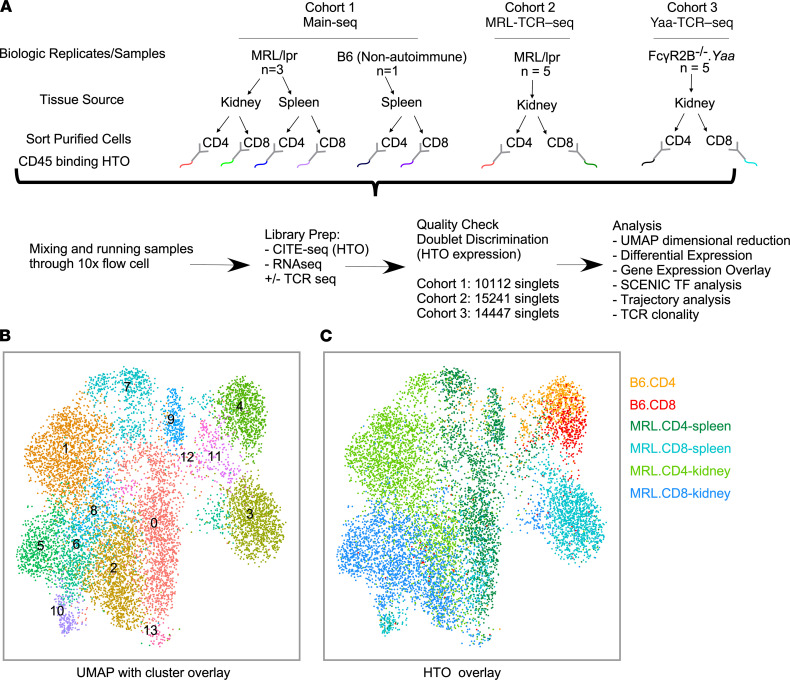
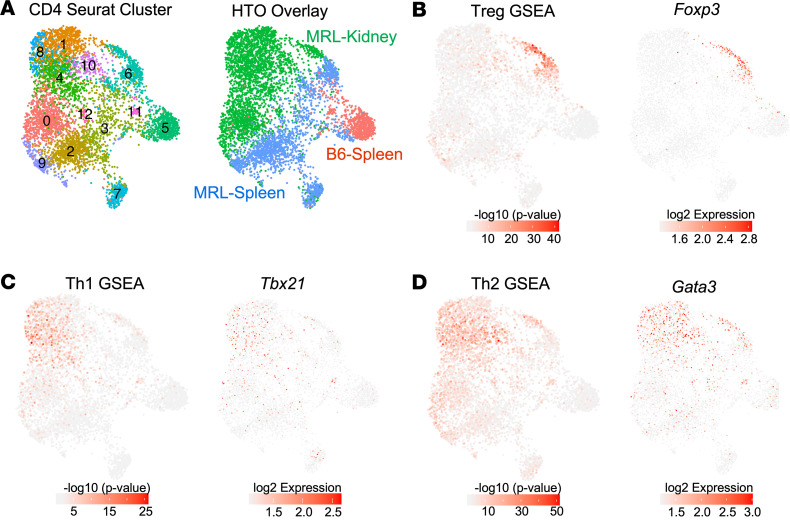
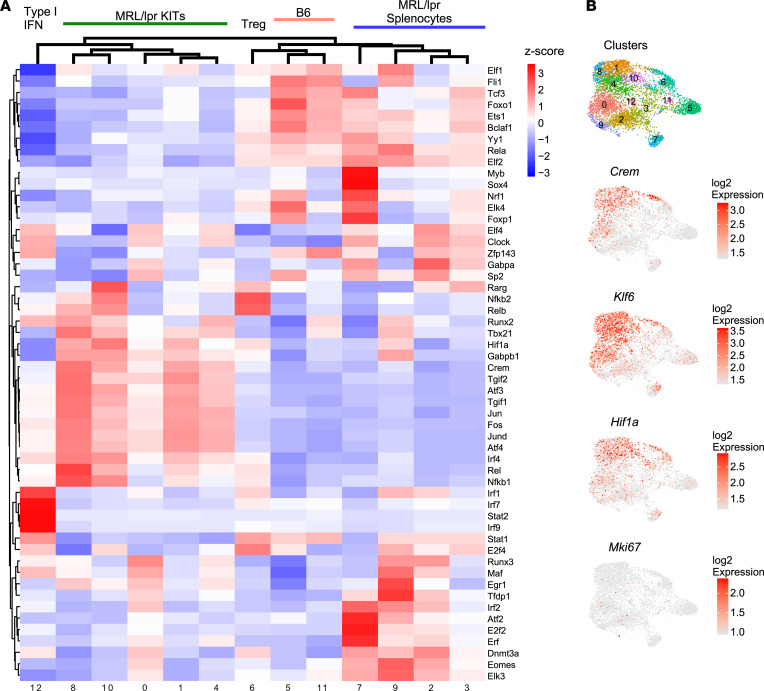
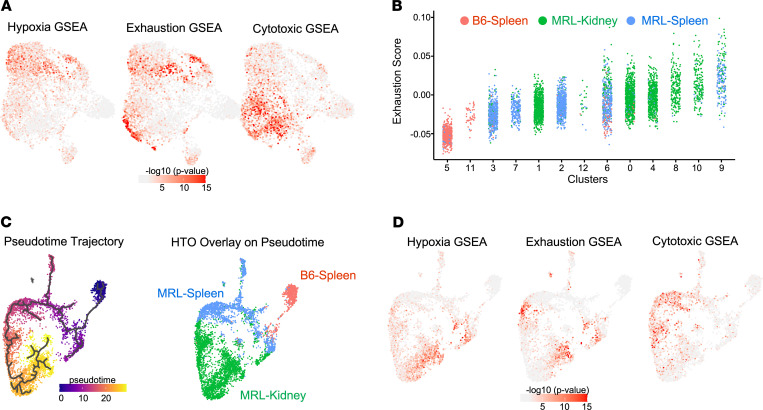
We previously found that kidney-infiltrating T cells (KITs) in murine lupus nephritis (LN) resembled dysfunctional T cells that infiltrate tumors. This unexpected finding raised the question of how to reconcile the "exhausted" phenotype of KITs with ongoing tissue destruction in LN. To address this, we performed single-cell RNA-Seq and TCR-Seq of KITs in murine lupus models. We found that CD8+ KITs existed first in a transitional state, before clonally expanding and evolving toward exhaustion. On the other hand, CD4+ KITs did not fit into current differentiation paradigms but included both hypoxic and cytotoxic subsets with a pervasive exhaustion signature. Thus, autoimmune nephritis is unlike acute pathogen immunity; rather, the kidney microenvironment suppresses T cells by progressively inducing exhausted states. Our findings suggest that LN, a chronic condition, results from slow evolution of damage caused by dysfunctional T cells and their precursors on the way to exhaustion. These findings have implications for both autoimmunity and tumor immunology.
Keywords: Autoimmune diseases; Autoimmunity; Immunology; T cells.
Conflict of interest statement
Figures
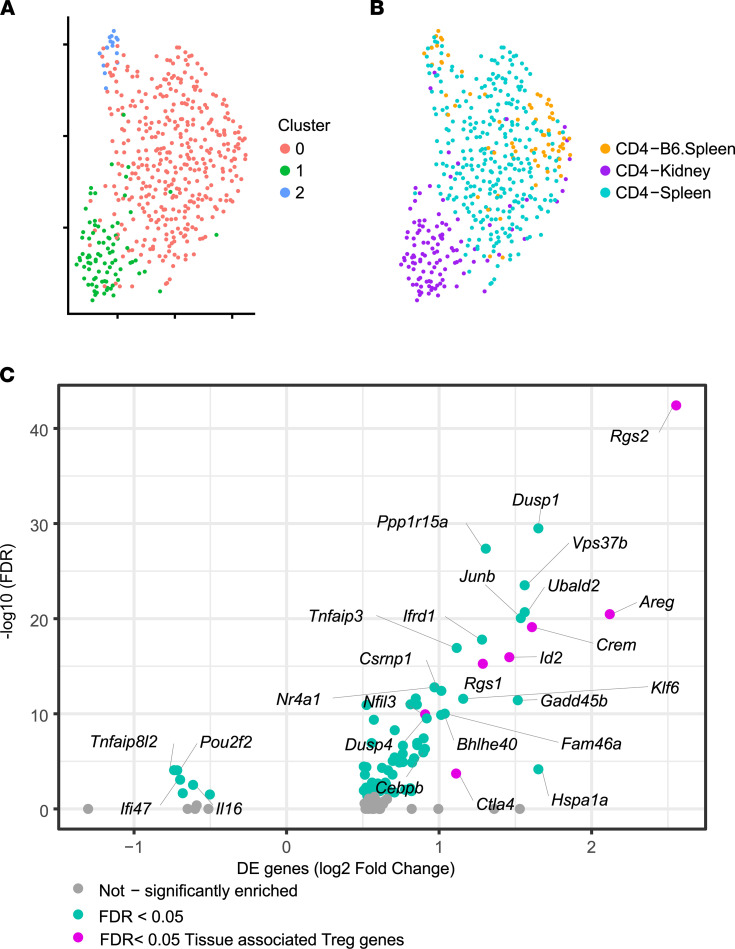
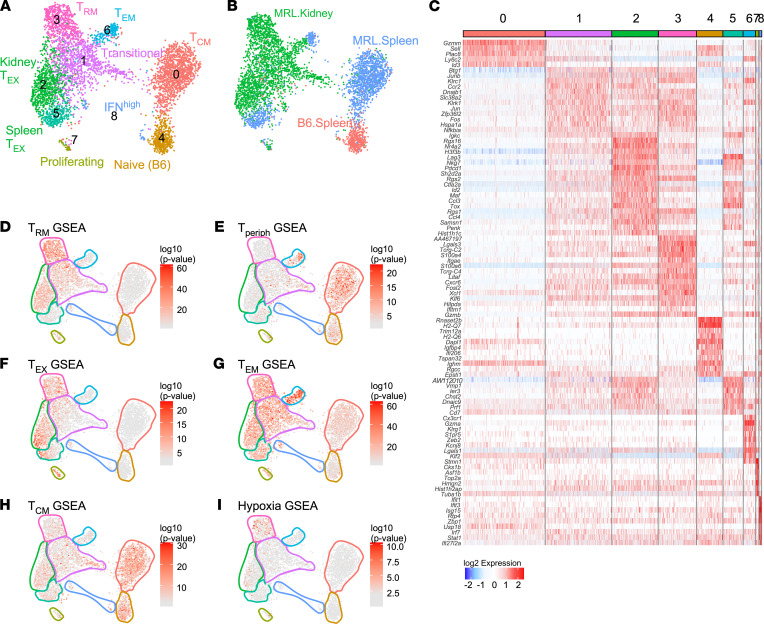
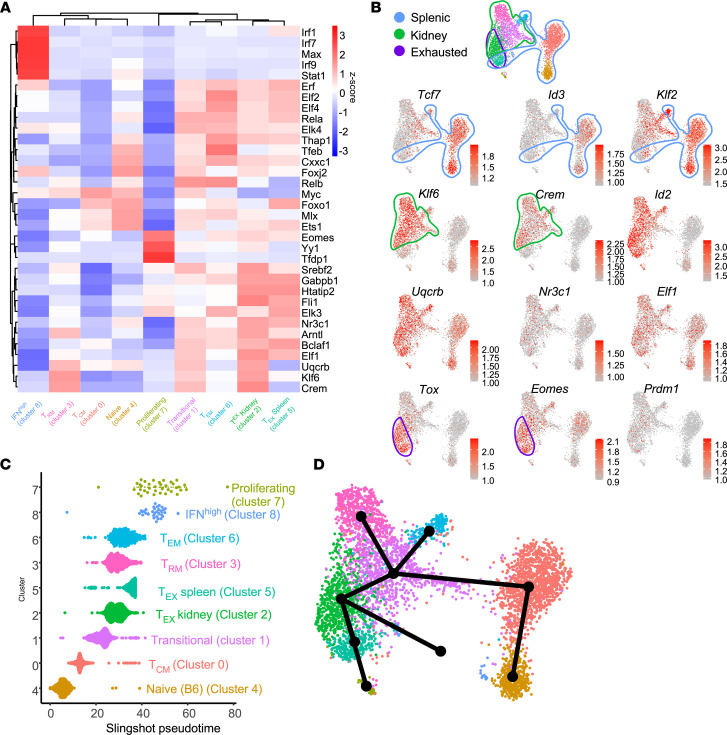
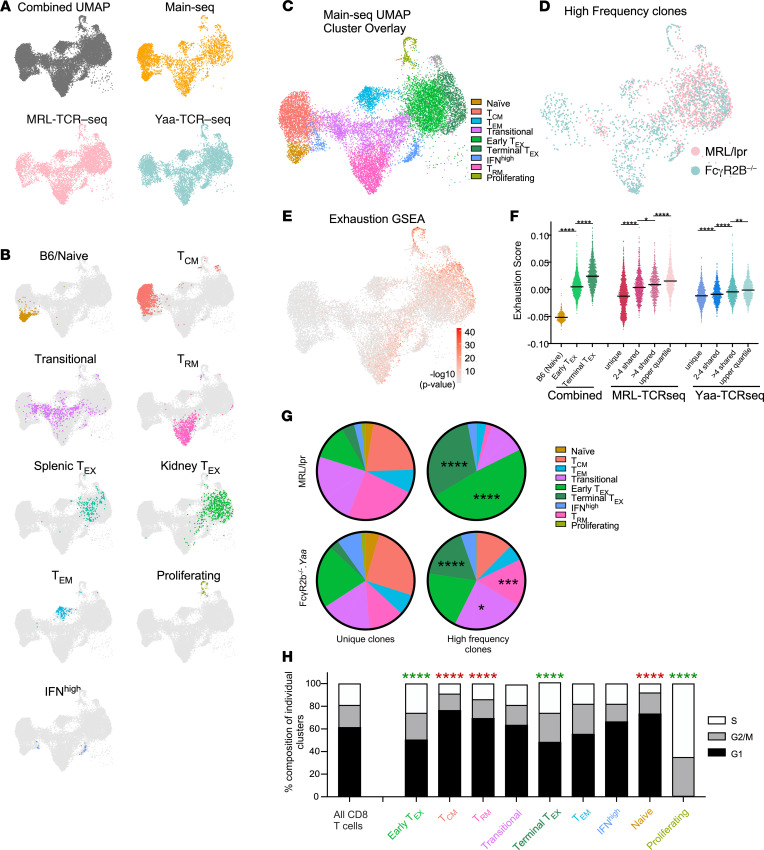
Similar articles
-
Kidney-infiltrating T cells in murine lupus nephritis are metabolically and functionally exhausted.J Clin Invest. 2018 Nov 1;128(11):4884-4897. doi: 10.1172/JCI120859. Epub 2018 Sep 24. J Clin Invest. 2018. PMID: 30130253 Free PMC article.
-
PD-1 activation mitigates lupus nephritis by suppressing hyperactive and heterogeneous PD-1+CD8+ T cells.Theranostics. 2025 Mar 31;15(11):5029-5044. doi: 10.7150/thno.107418. eCollection 2025. Theranostics. 2025. PMID: 40303350 Free PMC article.
-
Predominance of CD8+ T lymphocytes among periglomerular infiltrating cells and link to the prognosis of class III and class IV lupus nephritis.Arthritis Rheum. 2007 Jul;56(7):2362-70. doi: 10.1002/art.22654. Arthritis Rheum. 2007. PMID: 17599764
-
Update on Lupus Nephritis: Core Curriculum 2020.Am J Kidney Dis. 2020 Aug;76(2):265-281. doi: 10.1053/j.ajkd.2019.10.017. Epub 2020 Mar 24. Am J Kidney Dis. 2020. PMID: 32220510 Review.
-
Plasticity and heterogeneity of Th17 in immune-mediated kidney diseases.J Autoimmun. 2018 Feb;87:61-68. doi: 10.1016/j.jaut.2017.12.005. Epub 2017 Dec 21. J Autoimmun. 2018. PMID: 29275837 Review.
Cited by
-
T-cell exhaustion in immune-mediated inflammatory diseases: New implications for immunotherapy.Front Immunol. 2022 Sep 23;13:977394. doi: 10.3389/fimmu.2022.977394. eCollection 2022. Front Immunol. 2022. PMID: 36211414 Free PMC article. Review.
-
A current and future perspective on T cell receptor repertoire profiling.Front Genet. 2023 Jun 20;14:1159109. doi: 10.3389/fgene.2023.1159109. eCollection 2023. Front Genet. 2023. PMID: 37408774 Free PMC article. Review.
-
Pathogenic cellular and molecular mediators in lupus nephritis.Nat Rev Nephrol. 2023 Aug;19(8):491-508. doi: 10.1038/s41581-023-00722-z. Epub 2023 May 24. Nat Rev Nephrol. 2023. PMID: 37225921 Review.
-
Single-Cell Analysis Provides New Insights into the Roles of Tertiary Lymphoid Structures and Immune Cell Infiltration in Kidney Injury and Chronic Kidney Disease.Am J Pathol. 2025 Jan;195(1):40-54. doi: 10.1016/j.ajpath.2024.07.008. Epub 2024 Aug 7. Am J Pathol. 2025. PMID: 39097168 Free PMC article. Review.
-
Type I interferon drives T cell cytotoxicity by upregulation of interferon regulatory factor 7 in autoimmune kidney diseases in mice.Nat Commun. 2025 May 20;16(1):4686. doi: 10.1038/s41467-025-59819-7. Nat Commun. 2025. PMID: 40393992 Free PMC article.
References
-
- Hope HC, Salmond RJ. Targeting the tumor microenvironment and T cell metabolism for effective cancer immunotherapy. Eur J Immunol. 2019;49(8):1147–1152. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
