

Prediction of histone post-translational modification patterns based on nascent transcription data
- PMID: 35273399
- PMCID: PMC9444190
- DOI: 10.1038/s41588-022-01026-x
Prediction of histone post-translational modification patterns based on nascent transcription data
Abstract
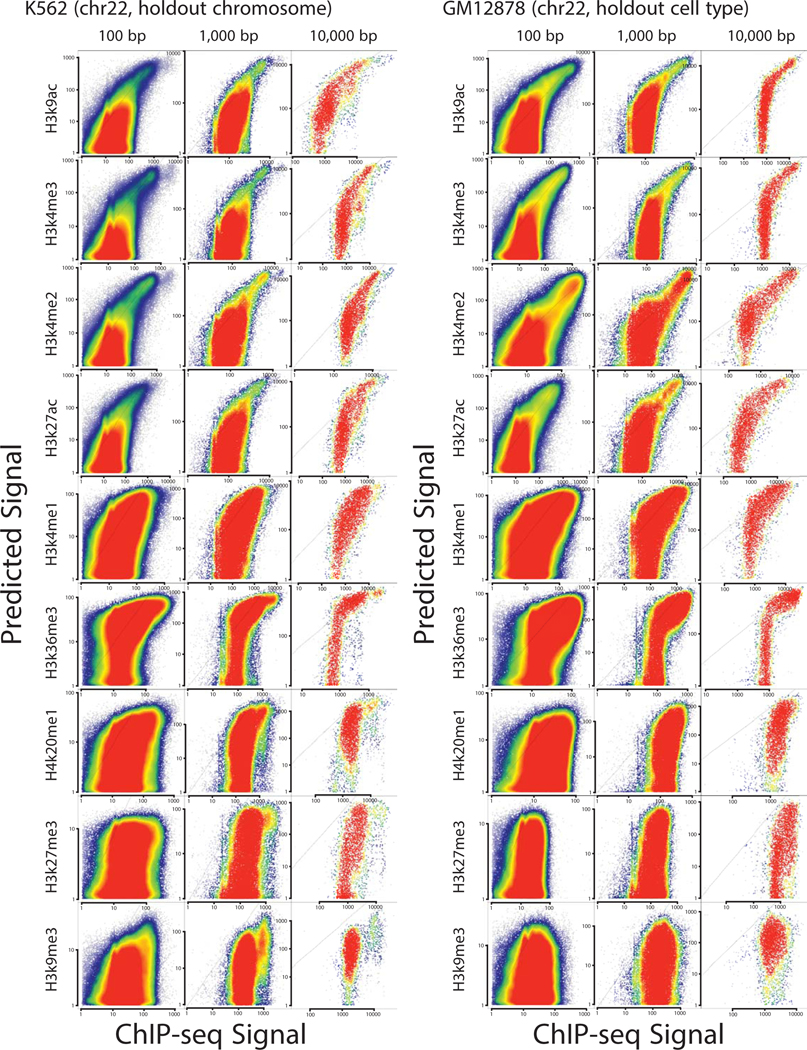
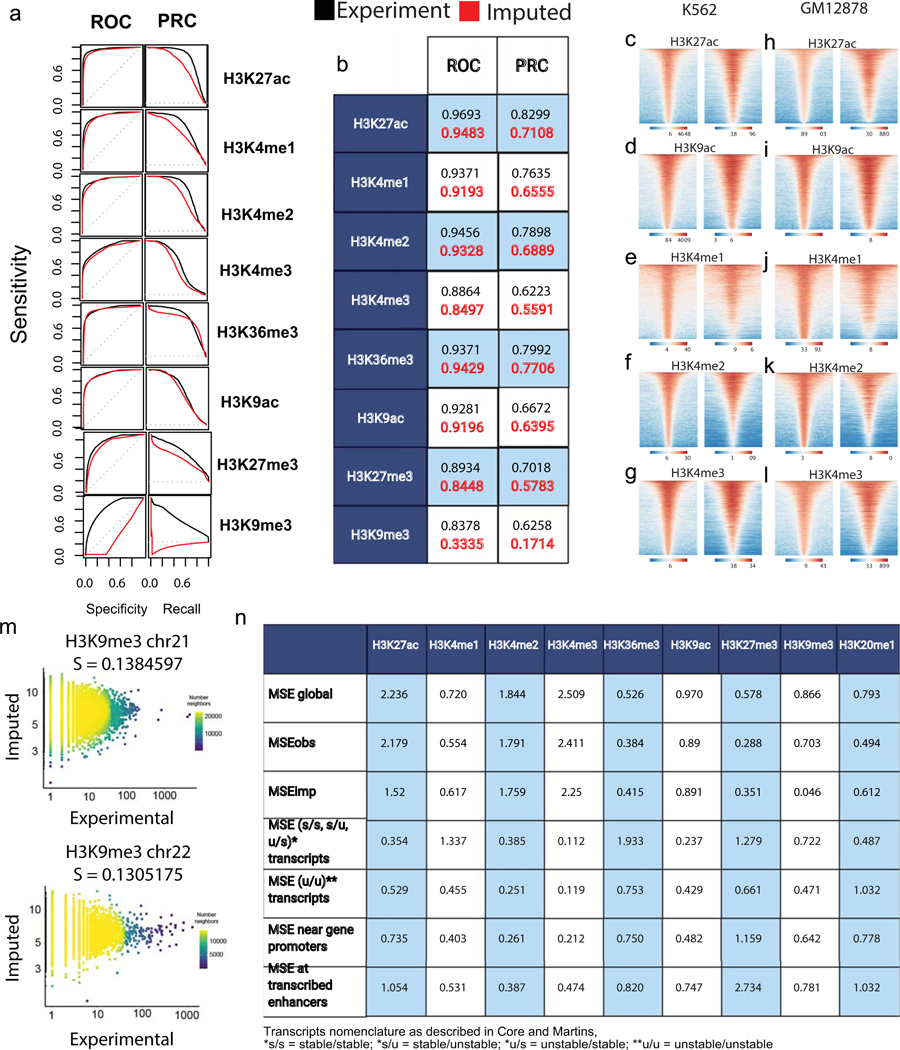
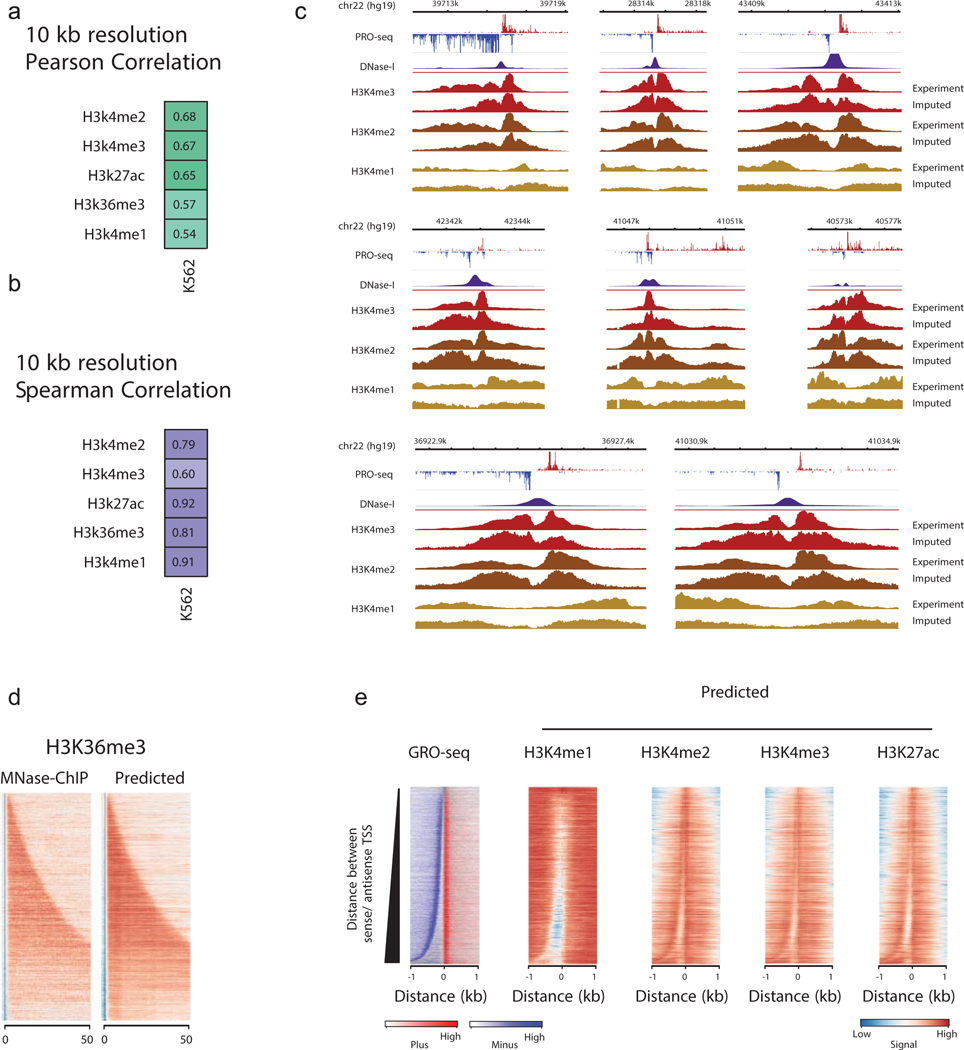
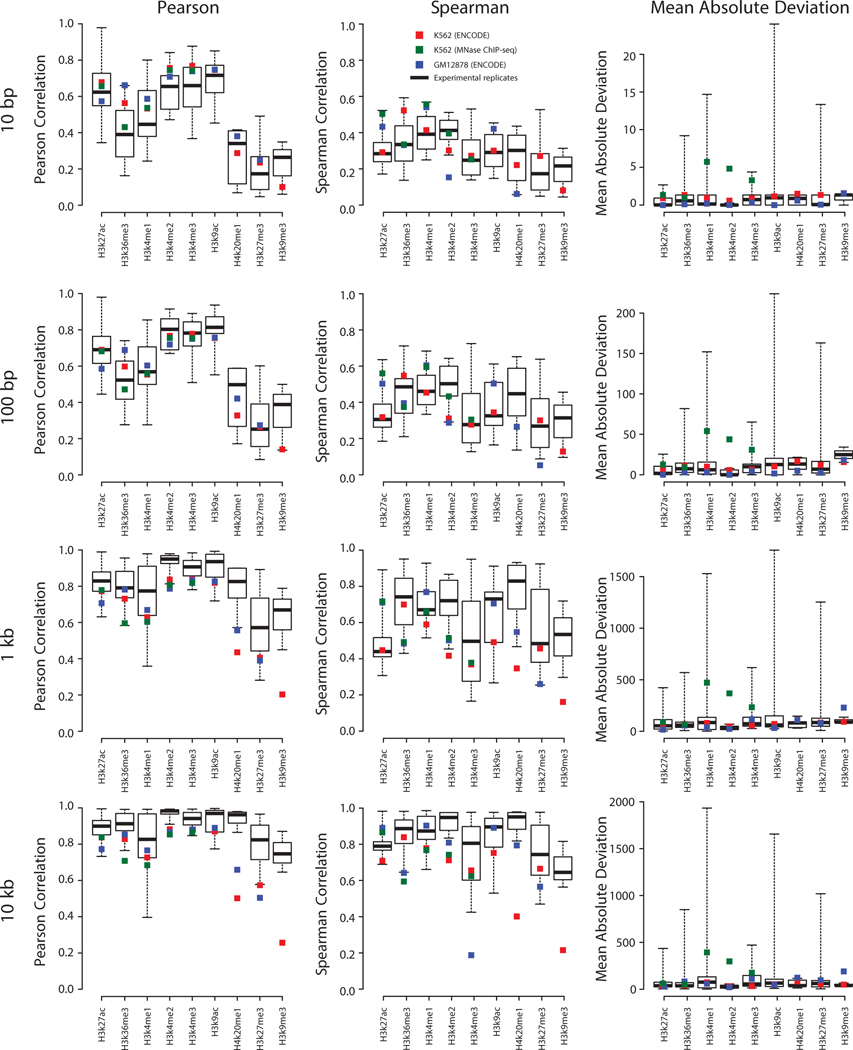
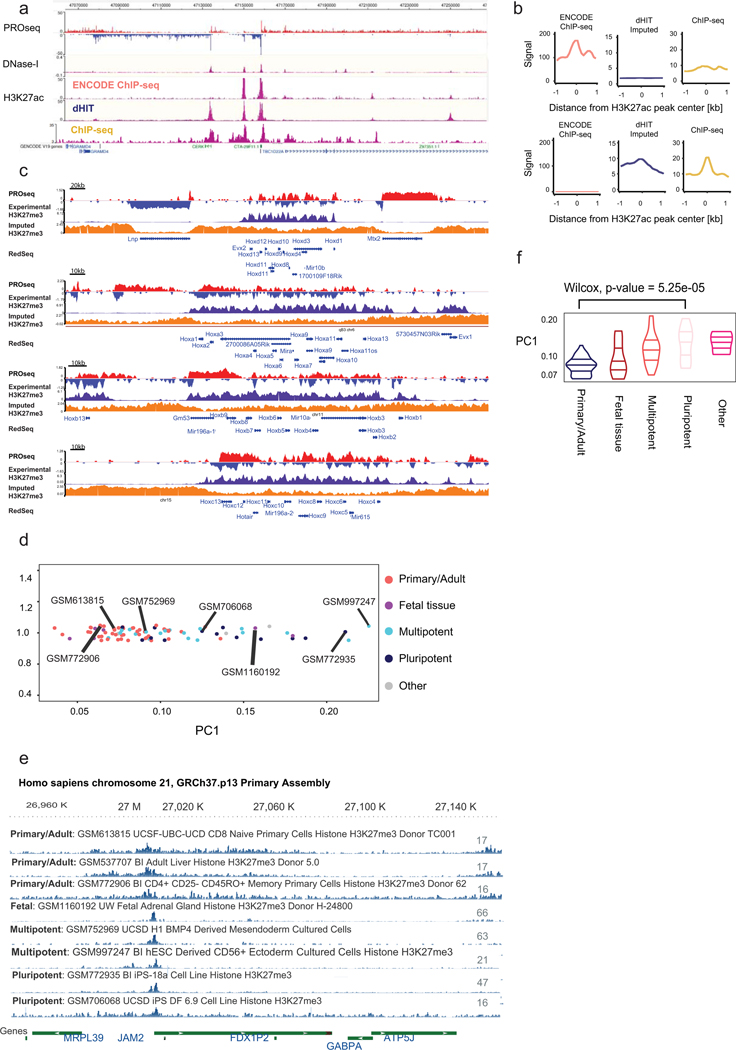
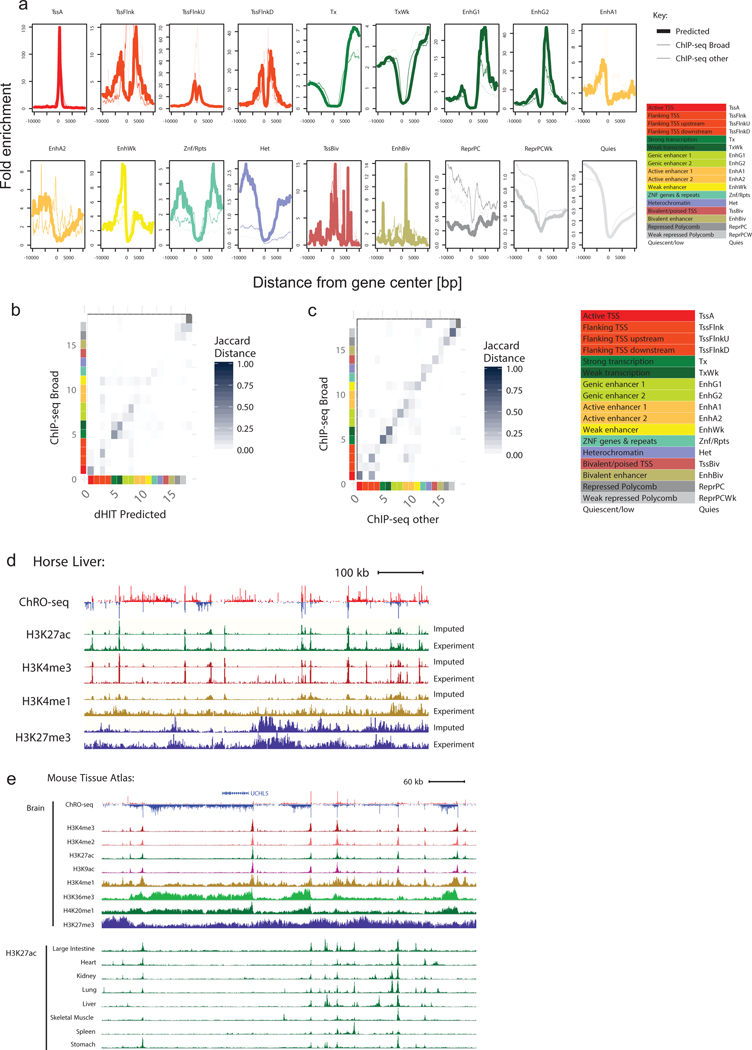
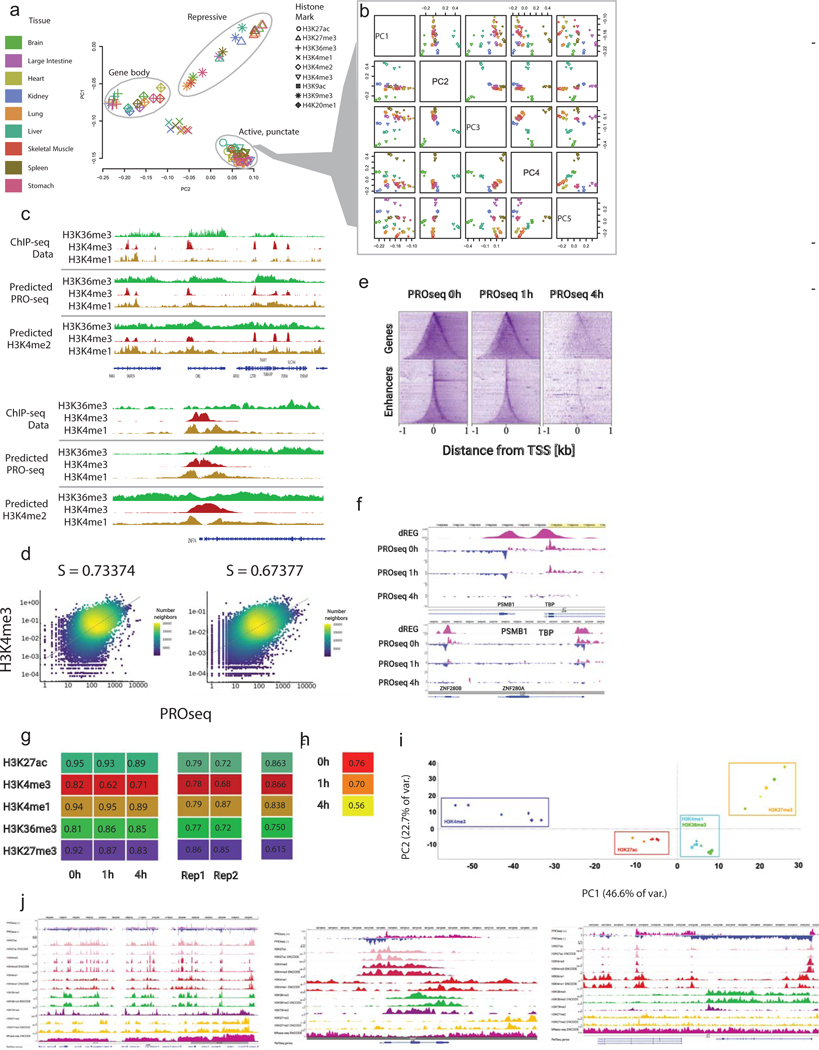
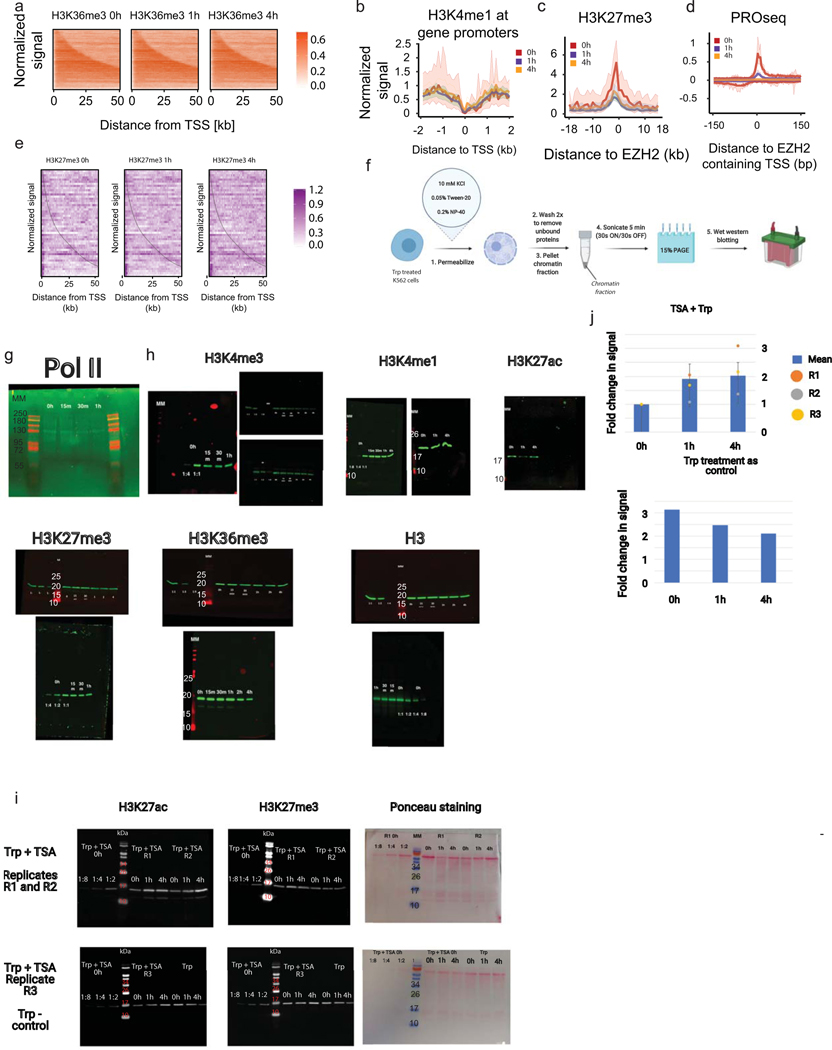
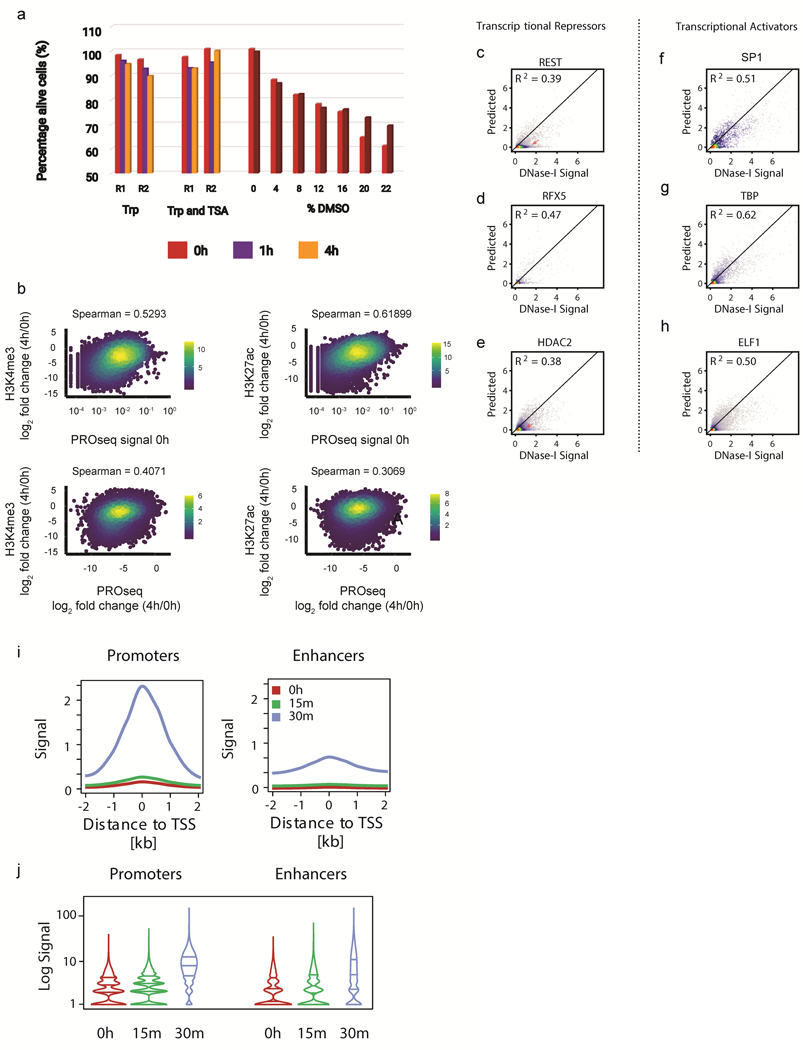
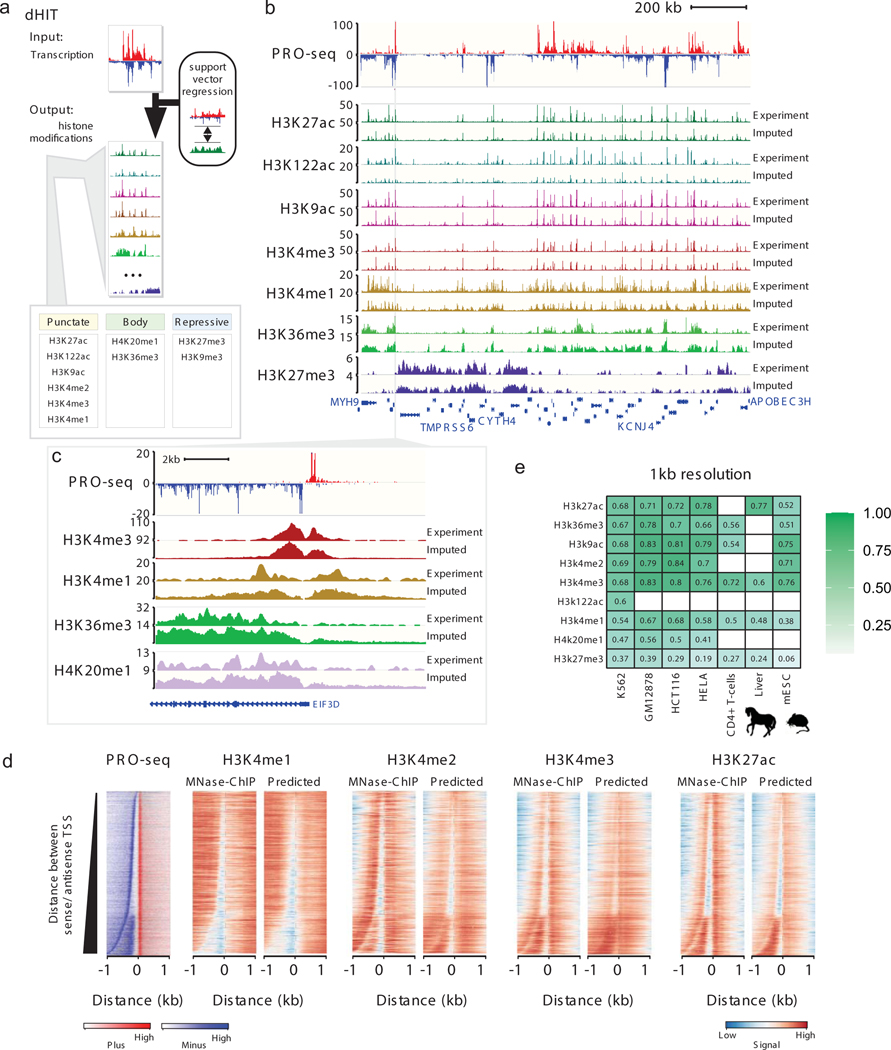
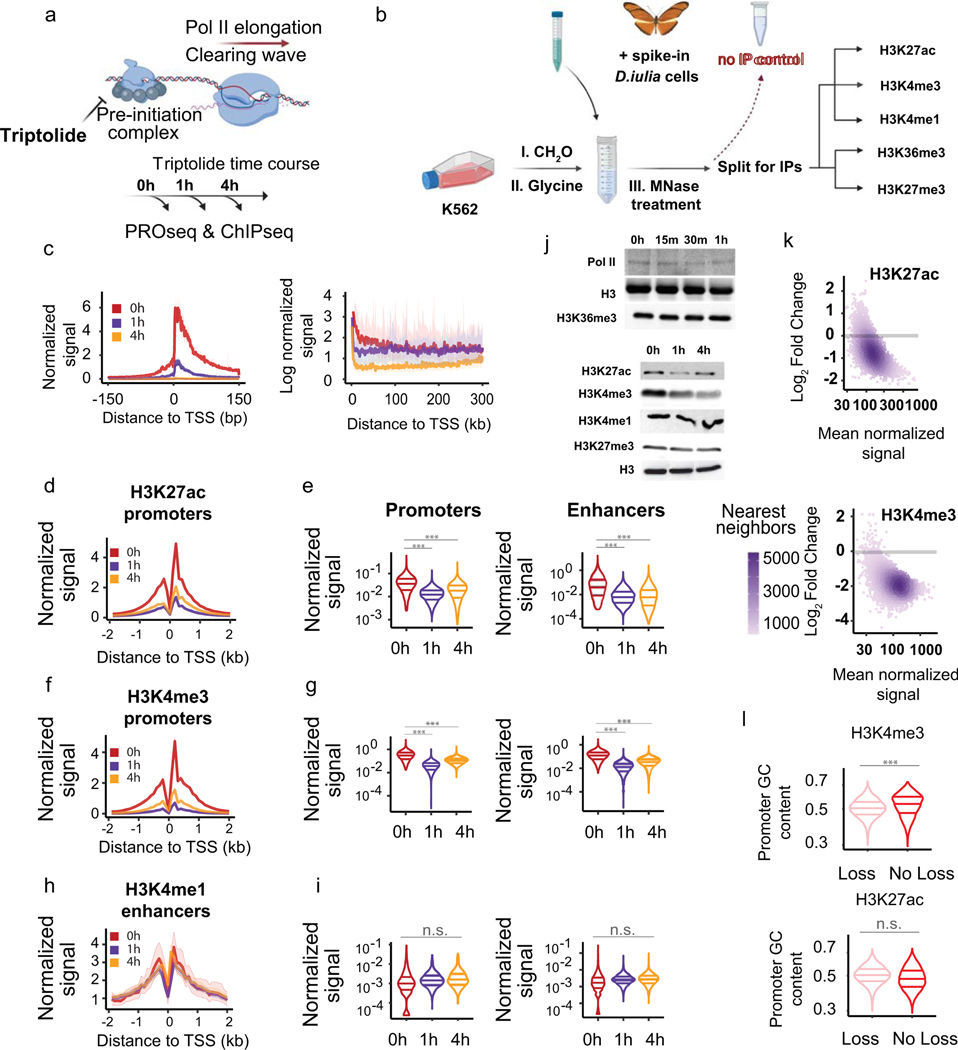
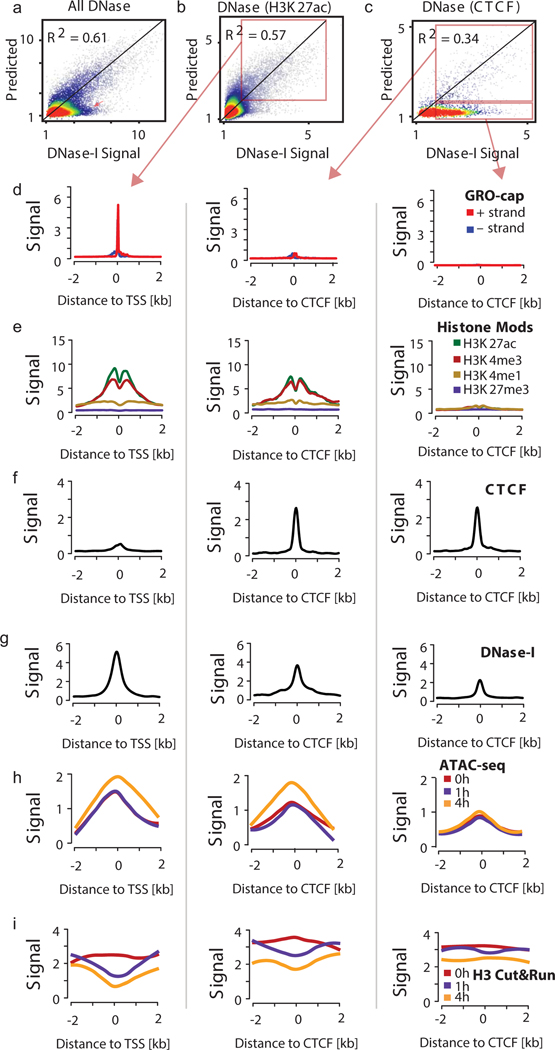
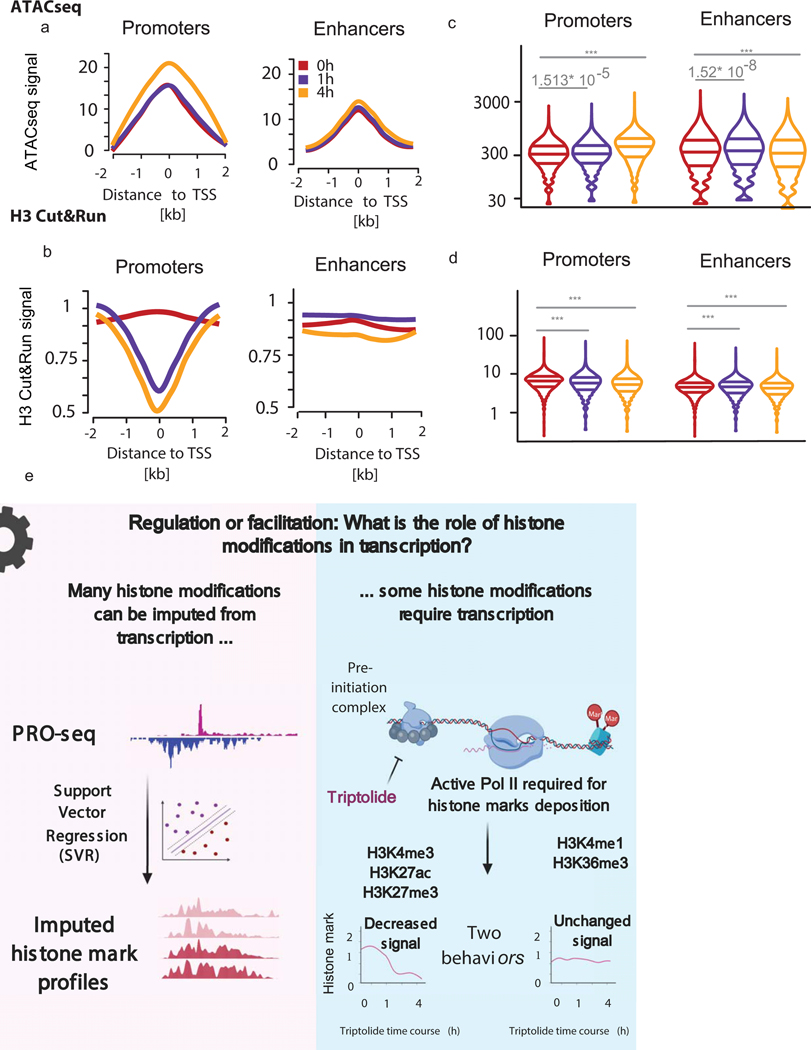
The role of histone modifications in transcription remains incompletely understood. Here, we examine the relationship between histone modifications and transcription using experimental perturbations combined with sensitive machine-learning tools. Transcription predicted the variation in active histone marks and complex chromatin states, like bivalent promoters, down to single-nucleosome resolution and at an accuracy that rivaled the correspondence between independent ChIP-seq experiments. Blocking transcription rapidly removed two punctate marks, H3K4me3 and H3K27ac, from chromatin indicating that transcription is required for active histone modifications. Transcription was also required for maintenance of H3K27me3, consistent with a role for RNA in recruiting PRC2. A subset of DNase-I-hypersensitive sites were refractory to prediction, precluding models where transcription initiates pervasively at any open chromatin. Our results, in combination with past literature, support a model in which active histone modifications serve a supportive, rather than an essential regulatory, role in transcription.
© 2022. The Author(s), under exclusive licence to Springer Nature America, Inc.
Conflict of interest statement
Competing Interests Statement
The authors declare no competing interests.
Figures



Comment in
-
Nascent transcription as a predictor and driver of histone modifications.Nat Genet. 2022 Mar;54(3):223-224. doi: 10.1038/s41588-021-01004-9. Nat Genet. 2022. PMID: 35273400 No abstract available.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
