

Congenital Adrenal Hyperplasia and Ehlers-Danlos Syndrome
- PMID: 35282436
- PMCID: PMC8913572
- DOI: 10.3389/fendo.2022.803226
Congenital Adrenal Hyperplasia and Ehlers-Danlos Syndrome
Abstract
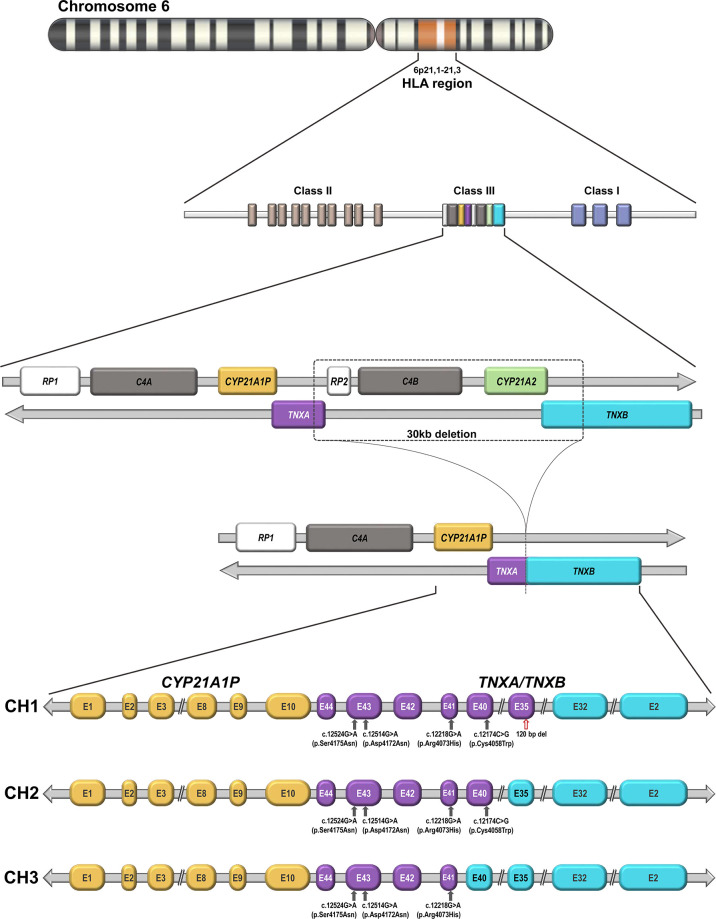
Congenital adrenal hyperplasia (CAH) secondary to 21-hydroxylase deficiency is an autosomal recessive disorder. The 21-hydroxylase enzyme P450c21 is encoded by the CYP21A2 gene located on chromosome 6p21.33 within the HLA major histocompatibility complex. This locus also contains the CYP21A1P, a non-functional pseudogene, that is highly homologous to the CYP21A2 gene. Other duplicated genes are C4A and C4B, that encode two isoforms of complement factor C4, the RP1 gene that encodes a serine/threonine protein kinase, and the TNXB gene that, encodes the extracellular matrix glycoprotein tenascin-X (TNX). TNX plays a role in collagen deposition by dermal fibroblasts and is expressed in the dermis of the skin and the connective tissue of the heart and skeletal muscle. During meiosis, misalignment may occur producing large gene deletions or gene conversion events resulting in chimeric genes. Chimeric recombination may occur between TNXB and TNXA. Three TNXA/TNXB chimeras have been described that differ in the junction site (CH1 to CH3) and result in a contiguous CYP21A2 and TNXB gene deletion, causing CAH-X syndrome. TNXB deficiency is associated with Ehlers Danlos syndrome (EDS). EDS comprises a clinically and genetically heterogeneous group of connective tissue disorders. As molecular analysis of the TNXB gene is challenging, the TNX-deficient type EDS is probably underdiagnosed. In this minireview, we will address the different strategies of molecular analysis of the TNXB-gene, as well as copy number variations and genetic status of TNXB in different cohorts. Furthermore, clinical features of EDS and clinical recommendations for long-term follow-up are discussed.
Keywords: CAH-X; CYP21A2; Ehlers-Danlos Syndrome; TNXB; congenital adrenal hyperplasia.
Copyright © 2022 Marino, Moresco, Perez Garrido, Ramirez and Belgorosky.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
References
-
- Yang Z, Mendoza AR, Welch TR, Zipf WB, Yu CY. Modular Variations of the Human Major Histocompatibility Complex Class III Genes for Serine/Threonine Kinase RP, Complement Component C4, Steroid 21-Hydroxylase CYP21, and Tenascin TNX (the RCCX Module). A Mechanism for Gene Deletions and Disease Associations. J Biol Chem (1999) 274(17):12147–56. doi: 10.1074/jbc.274.17.12147 - DOI - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous