

Hunter Syndrome: The Phenotype of a Rare Storage Disease
- PMID: 35282545
- PMCID: PMC8906563
- DOI: 10.7759/cureus.21985
Hunter Syndrome: The Phenotype of a Rare Storage Disease
Abstract
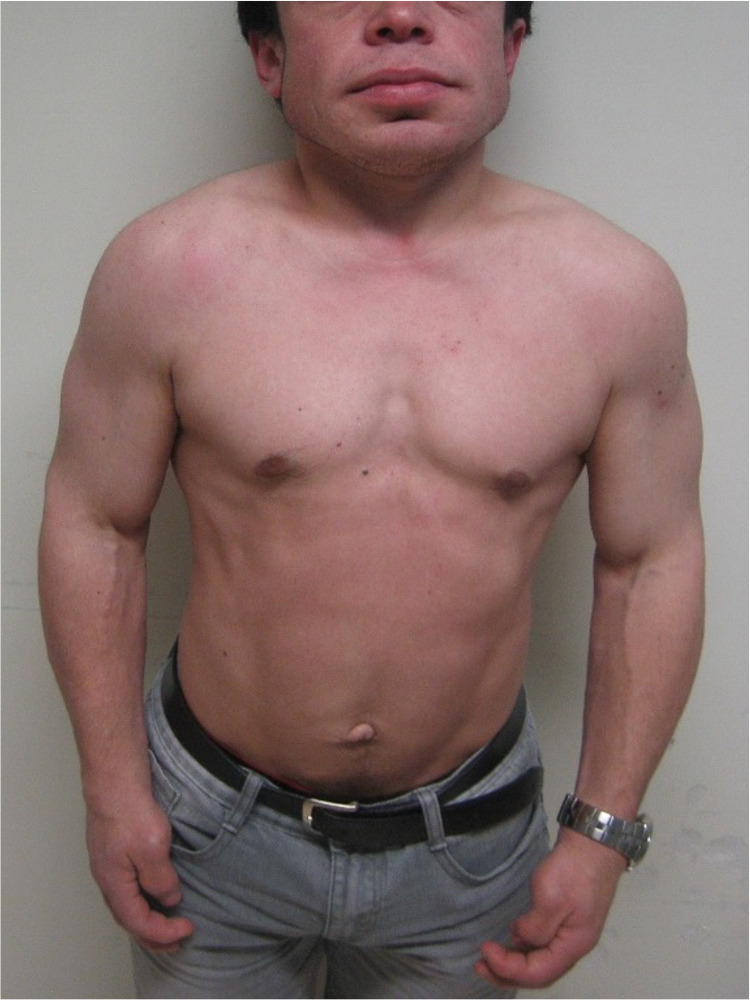
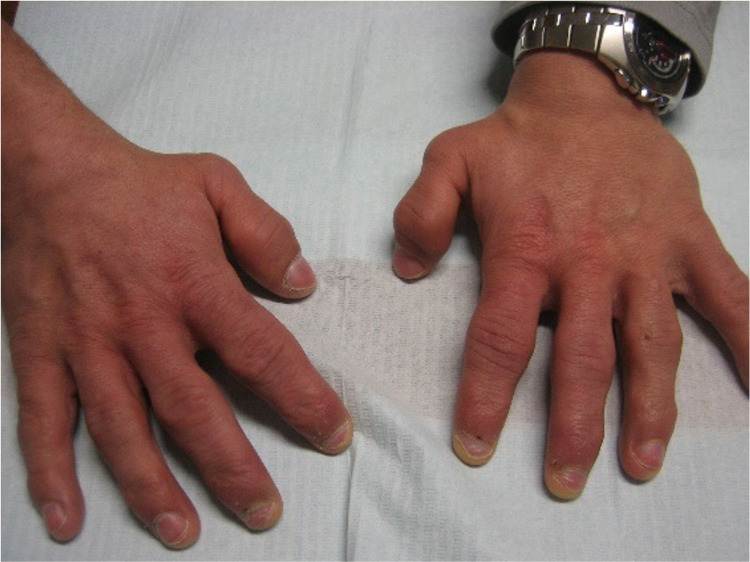
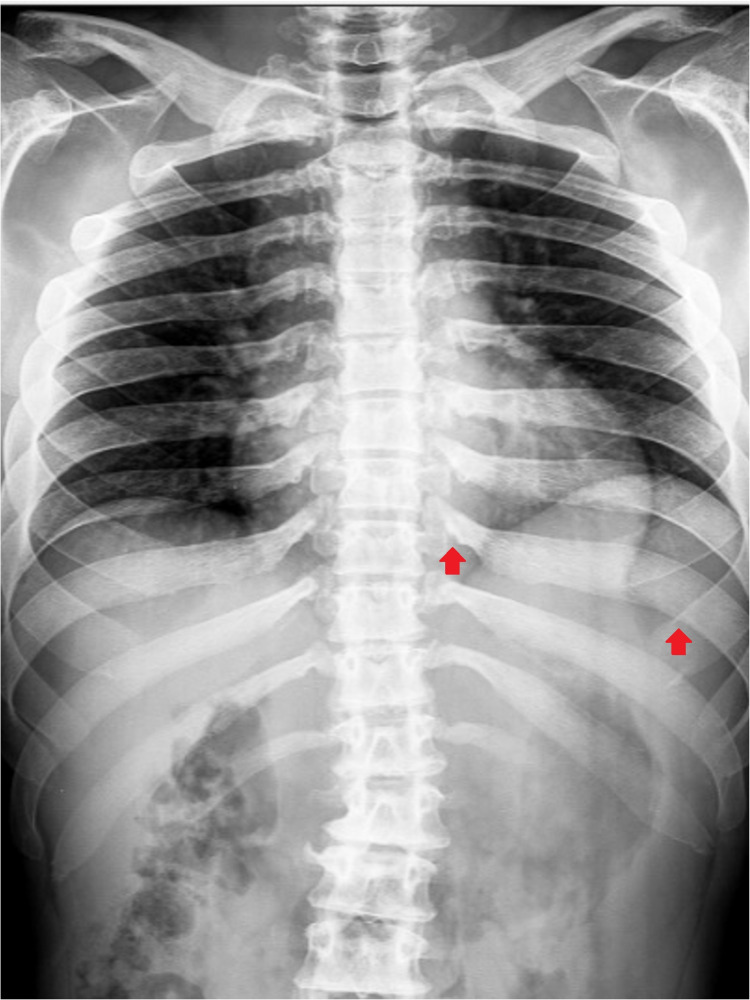
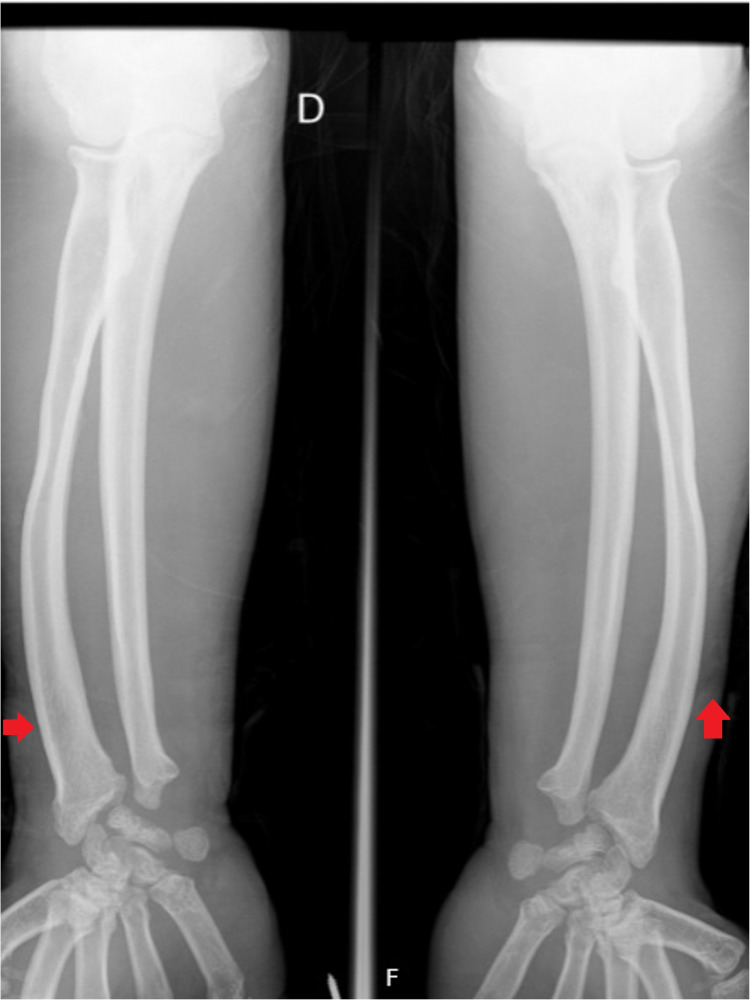
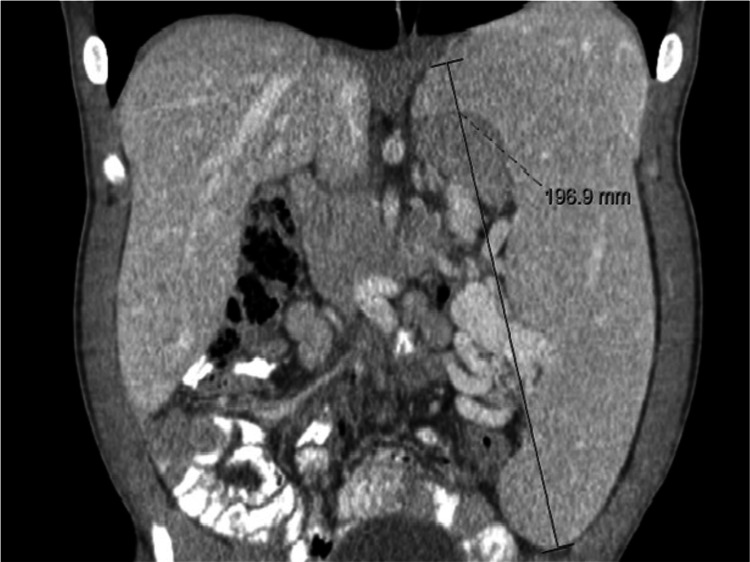
Hunter syndrome is a rare lysosomal storage disorder with systemic involvement that occurs over time. Affected patients have coarse facial features, growth retardation with short stature, and skeletal deformities called dysostosis multiplex; joint stiffness, progressive mental retardation, and organomegaly are some of the clinical signs. It ranges from mild to severe manifestations and the distinction between them is related to neurological involvement. Cardiac and respiratory failure is commonly the cause of early death (before adulthood) for severe forms, but those with attenuated forms who have normal cognitive development can survive until late adulthood. Treatment with enzyme replacement therapy is available and can improve the prognosis of this disease. The authors present a case of a 36-year-old male with Hunter syndrome to show not only the clinical features typical of this multisystemic disease that should alert to a prompt investigation but also to remind that treatment must start as early as possible to reach the best outcome. Management of this disease is typically challenging and requires a multidisciplinary approach.
Keywords: dysostosis multiplex; hunter syndrome; inborn errors of metabolism; mucopolysaccharidosis; rare diseases.
Copyright © 2022, Sousa Martins et al.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
References
-
- Orphanet. Mucopolysaccharidosis type 2. [ Dec; 2021 ];https://www.orpha.net/consor/cgi-bin/OC_Exp.php?lng=en&Expert=580 2019
-
- Mucopolysaccharidosis type II (Hunter syndrome): clinical and biochemical aspects of the disease and approaches to its diagnosis and treatment. Mohamed S, He QQ, Singh AA, Ferro V. Adv Carbohydr Chem Biochem. 2020;77:71–117. - PubMed
Publication types
LinkOut - more resources
Full Text Sources