

The Molecular Brakes of Adipose Tissue Lipolysis
- PMID: 35283787
- PMCID: PMC8907745
- DOI: 10.3389/fphys.2022.826314
The Molecular Brakes of Adipose Tissue Lipolysis
Abstract
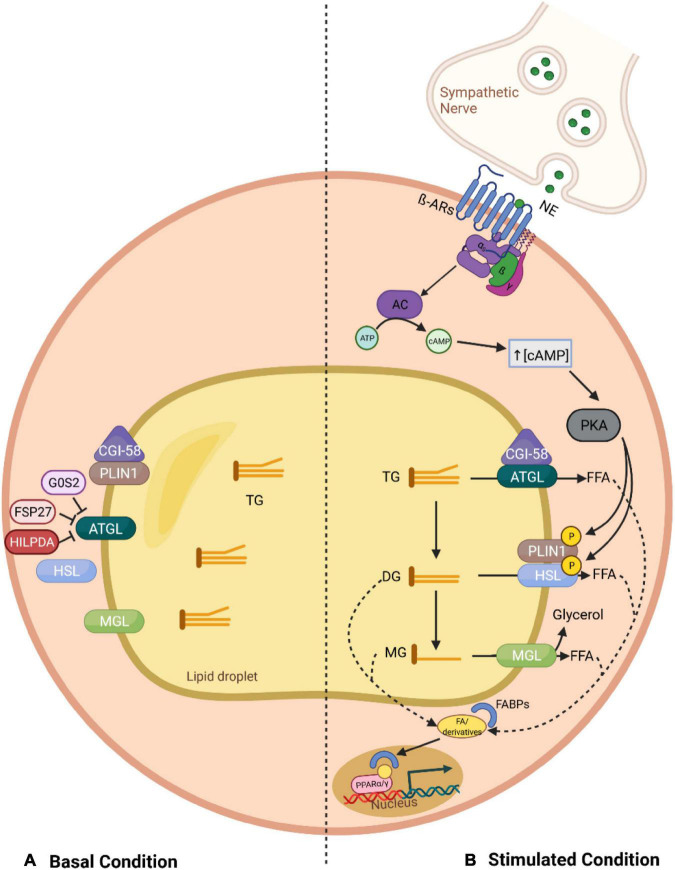
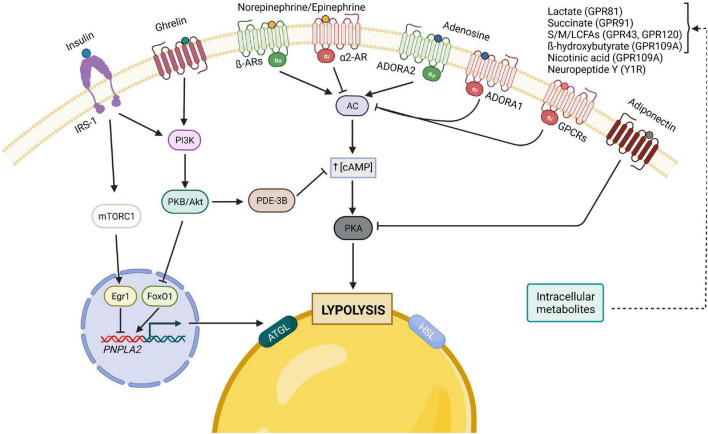
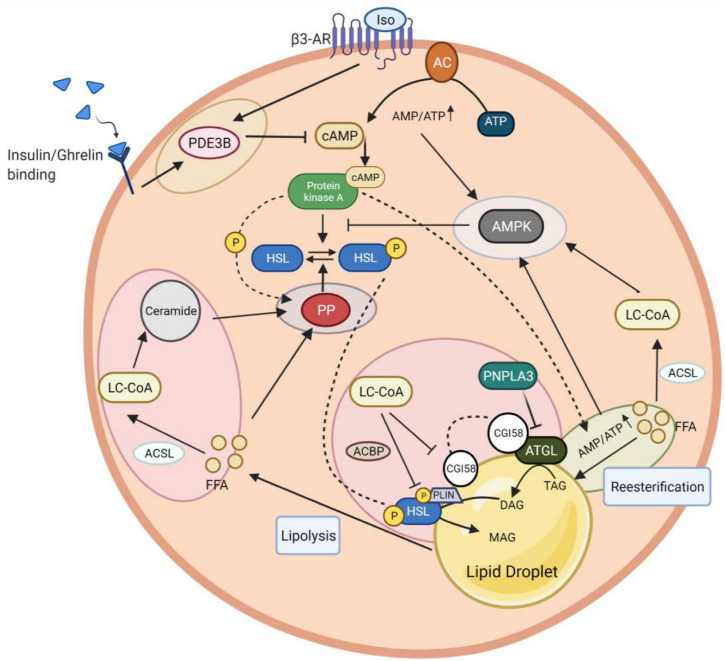
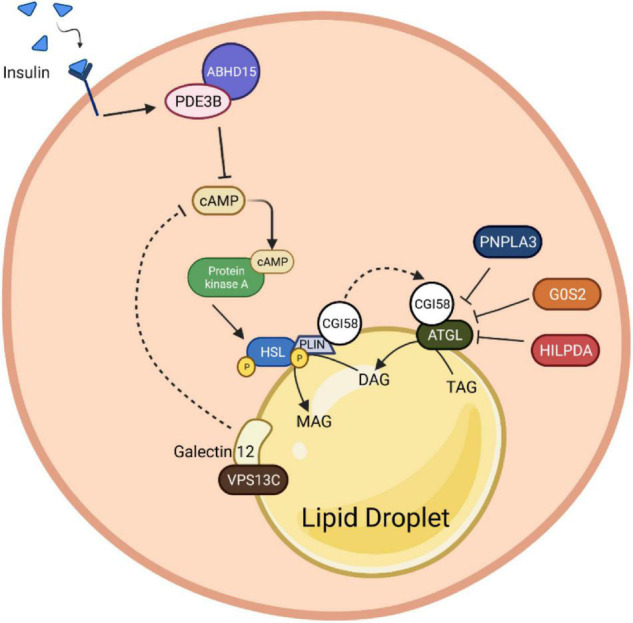
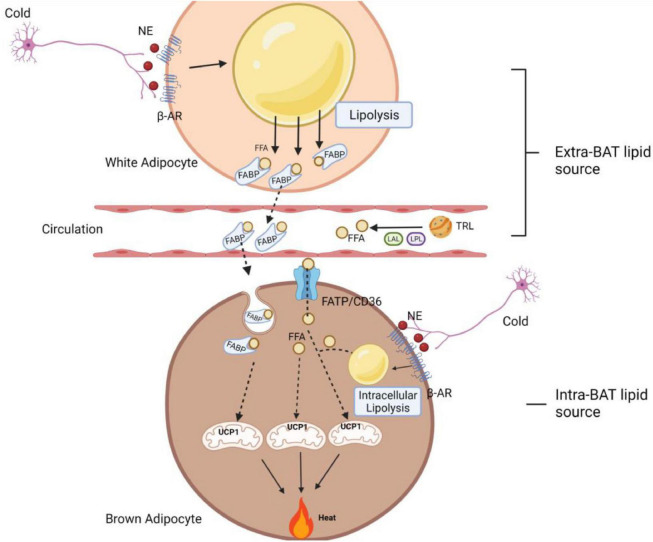
Adaptation to changes in energy availability is pivotal for the survival of animals. Adipose tissue, the body's largest reservoir of energy and a major source of metabolic fuel, exerts a buffering function for fluctuations in nutrient availability. This functional plasticity ranges from energy storage in the form of triglycerides during periods of excess energy intake to energy mobilization via lipolysis in the form of free fatty acids for other organs during states of energy demands. The subtle balance between energy storage and mobilization is important for whole-body energy homeostasis; its disruption has been implicated as contributing to the development of insulin resistance, type 2 diabetes and cancer cachexia. As a result, adipocyte lipolysis is tightly regulated by complex regulatory mechanisms involving lipases and hormonal and biochemical signals that have opposing effects. In thermogenic brown and brite adipocytes, lipolysis stimulation is the canonical way for the activation of non-shivering thermogenesis. Lipolysis proceeds in an orderly and delicately regulated manner, with stimulation through cell-surface receptors via neurotransmitters, hormones, and autocrine/paracrine factors that activate various intracellular signal transduction pathways and increase kinase activity. The subsequent phosphorylation of perilipins, lipases, and cofactors initiates the translocation of key lipases from the cytoplasm to lipid droplets and enables protein-protein interactions to assemble the lipolytic machinery on the scaffolding perilipins at the surface of lipid droplets. Although activation of lipolysis has been well studied, the feedback fine-tuning is less well appreciated. This review focuses on the molecular brakes of lipolysis and discusses some of the divergent fine-tuning strategies in the negative feedback regulation of lipolysis, including delicate negative feedback loops, intermediary lipid metabolites-mediated allosteric regulation and dynamic protein-protein interactions. As aberrant adipocyte lipolysis is involved in various metabolic diseases and releasing the brakes on lipolysis in thermogenic adipocytes may activate thermogenesis, targeting adipocyte lipolysis is thus of therapeutic interest.
Keywords: adipocytes; feedback mechanisms; free fatty acid; lipolysis; lipophagy; molecular brakes; reesterification; thermogenesis.
Copyright © 2022 Li, Li, Ngandiri, Llerins Perez, Wolf and Wang.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
References
-
- Ahmad F., Lindh R., Tang Y., Weston M., Degerman E., Manganiello V. C. (2007). Insulin-induced formation of macromolecular complexes involved in activation of cyclic nucleotide phosphodiesterase 3B (PDE3B) and its interaction with PKB. Biochem. J. 404 257–268. 10.1042/BJ20060960 - DOI - PMC - PubMed
Publication types
LinkOut - more resources
Full Text Sources