

DNAJB2-related Charcot-Marie-Tooth disease type 2: Pathomechanism insights and phenotypic spectrum widening
- PMID: 35286755
- PMCID: PMC9314055
- DOI: 10.1111/ene.15326
DNAJB2-related Charcot-Marie-Tooth disease type 2: Pathomechanism insights and phenotypic spectrum widening
Abstract
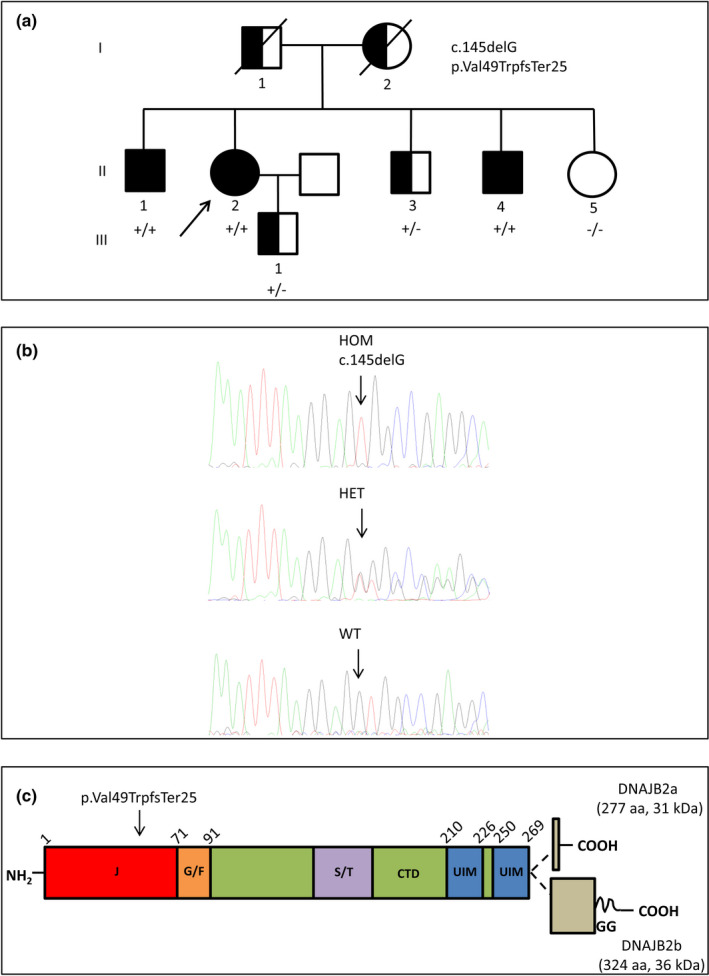
Background and purpose: Mutations in DNAJB2 are associated with autosomal recessive hereditary motor neuropathies/ Charcot-Marie-Tooth disease type 2 (CMT2). We describe an Italian family with CMT2 due to a homozygous DNAJB2 mutation and provide insight into the pathomechanisms.
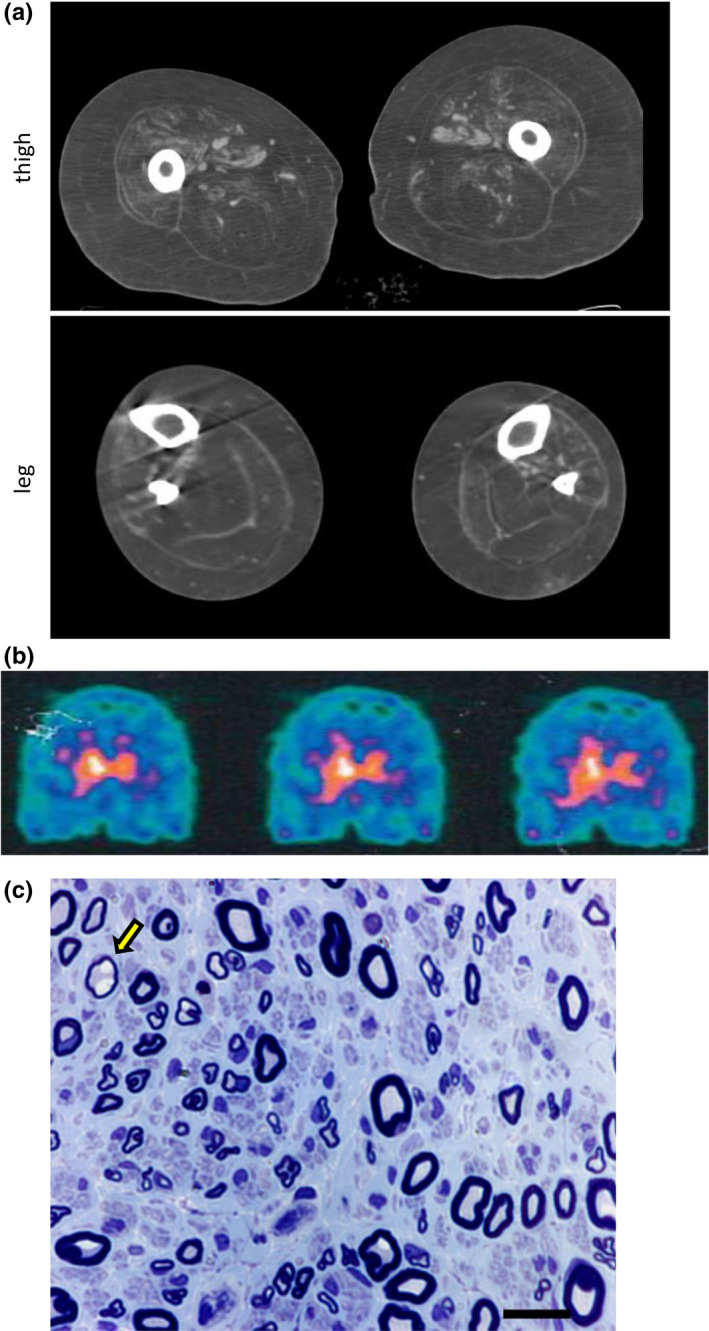
Methods: Patients with DNAJB2 mutations were characterized clinically, electrophysiologically and by means of skin biopsy. mRNA and protein levels were studied in lymphoblastoid cells (LCLs) from patients and controls.
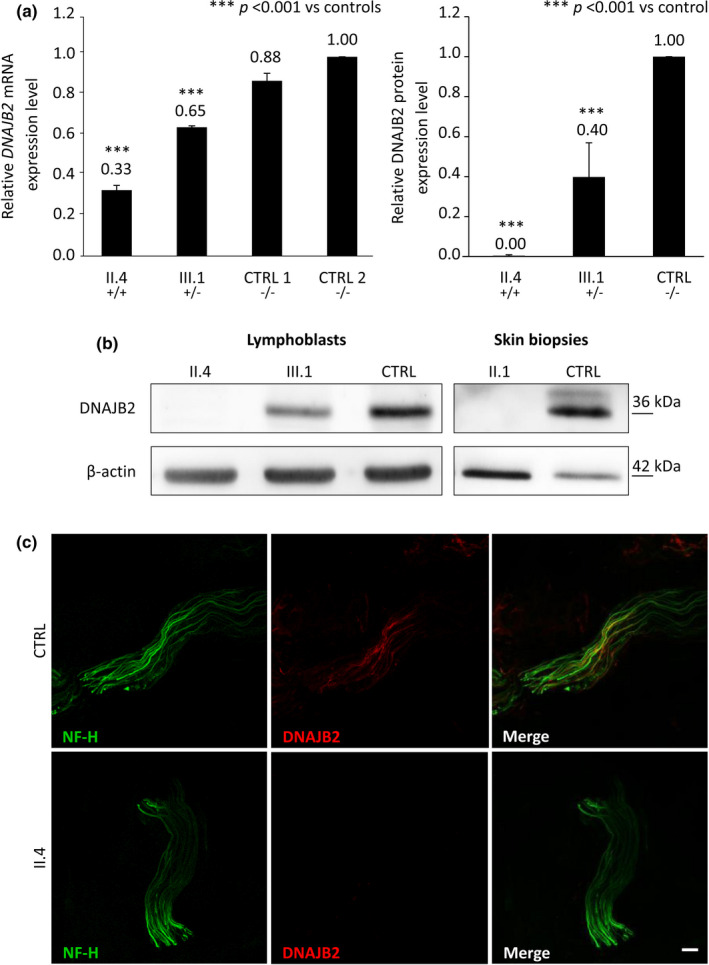
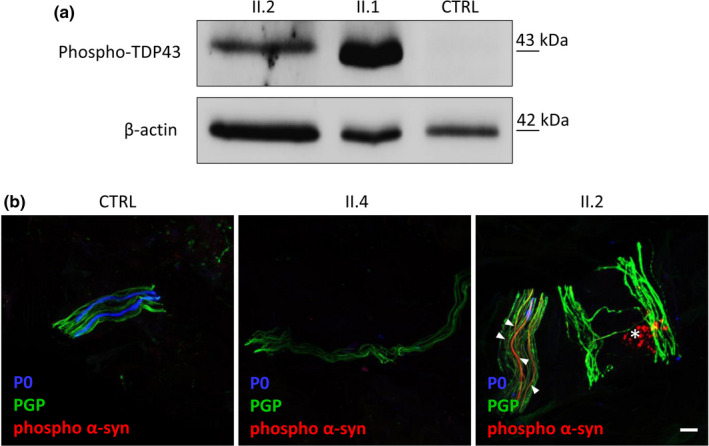
Results: Three affected siblings were found to carry a homozygous DNAJB2 null mutation segregating with the disease. The disease manifested in the second to third decade of life. Clinical examination showed severe weakness of the thigh muscles and complete loss of movement in the foot and leg muscles. Sensation was reduced in the lower limbs. All patients had severe hearing loss and the proband also had Parkinson's disease (PD). Nerve conduction studies showed an axonal motor and sensory length-dependent polyneuropathy. DNAJB2 expression studies revealed reduced mRNA levels and the absence of the protein in the homozygous subject in both LCLs and skin biopsy. Interestingly, we detected phospho-alpha-synuclein deposits in the proband, as already seen in PD patients, and demonstrated TDP-43 accumulation in patients' skin.
Conclusions: Our results broaden the clinical spectrum of DNAJB2-related neuropathies and provide evidence that DNAJB2 mutations should be taken into account as another causative gene of CMT2 with hearing loss and parkinsonism. The mutation likely acts through a loss-of-function mechanism, leading to toxic protein aggregation such as TDP-43. The associated parkinsonism resembles the classic PD form with the addition of abnormal accumulation of phospho-alpha-synuclein.
Keywords: Parkinson's disease; genetic and inherited disorders; neuropathology; peripheral neuropathies.
© 2022 The Authors. European Journal of Neurology published by John Wiley & Sons Ltd on behalf of European Academy of Neurology.
Conflict of interest statement
The authors have nothing to report.
Figures
References
-
- Pisciotta C, Shy ME. Neuropathy. Handb Clin Neurol. 2018;148:653‐665. - PubMed
-
- Previtali SC, Zhao E, Lazarevic D, et al. Expanding the spectrum of genes responsible for hereditary motor neuropathies. J Neurol Neurosurg Psychiatry. 2019;90:1171‐1179. - PubMed
-
- Blumen SC, Astord S, Robin V, et al. A rare recessive distal hereditary motor neuropathy with HSJ1 chaperone mutation. Ann Neurol. 2012;71:509‐519. - PubMed
-
- Gess B, Auer‐Grumbach M, Schirmacher A, et al. HSJ1‐related hereditary neuropathies: novel mutations and extended clinical spectrum. Neurology. 2014;83:1726‐1732. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
