

Theranostic F-SLOH mitigates Alzheimer's disease pathology involving TFEB and ameliorates cognitive functions in Alzheimer's disease models
- PMID: 35286997
- PMCID: PMC8921490
- DOI: 10.1016/j.redox.2022.102280
Theranostic F-SLOH mitigates Alzheimer's disease pathology involving TFEB and ameliorates cognitive functions in Alzheimer's disease models
Abstract
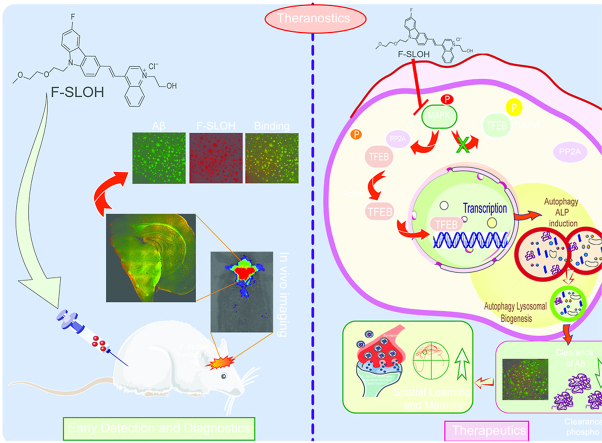
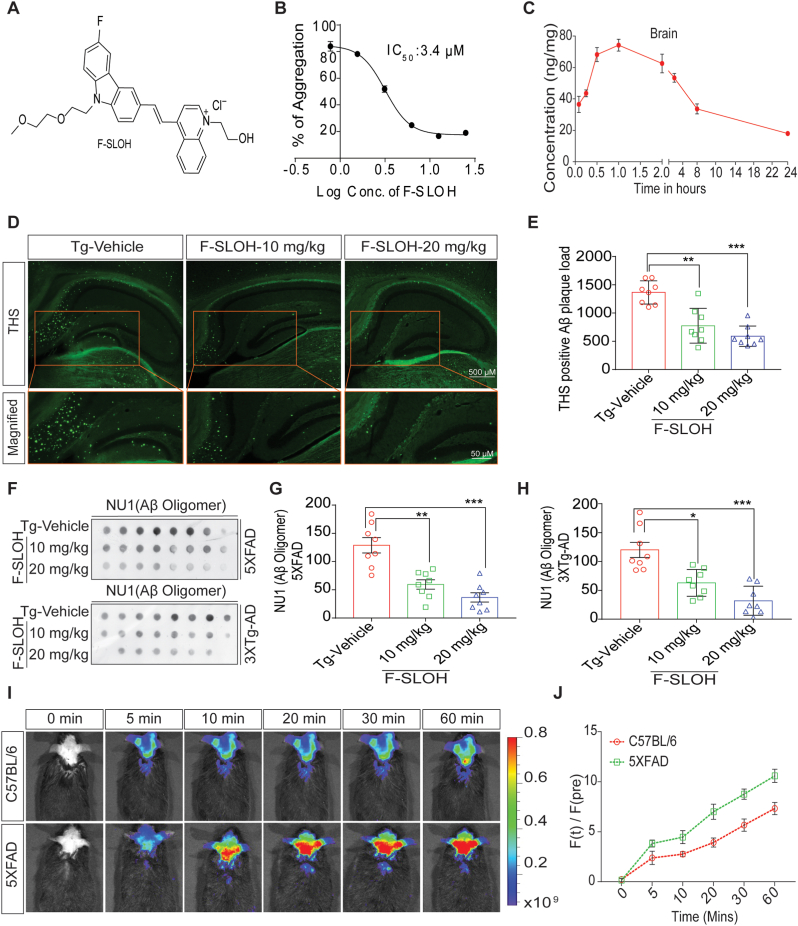
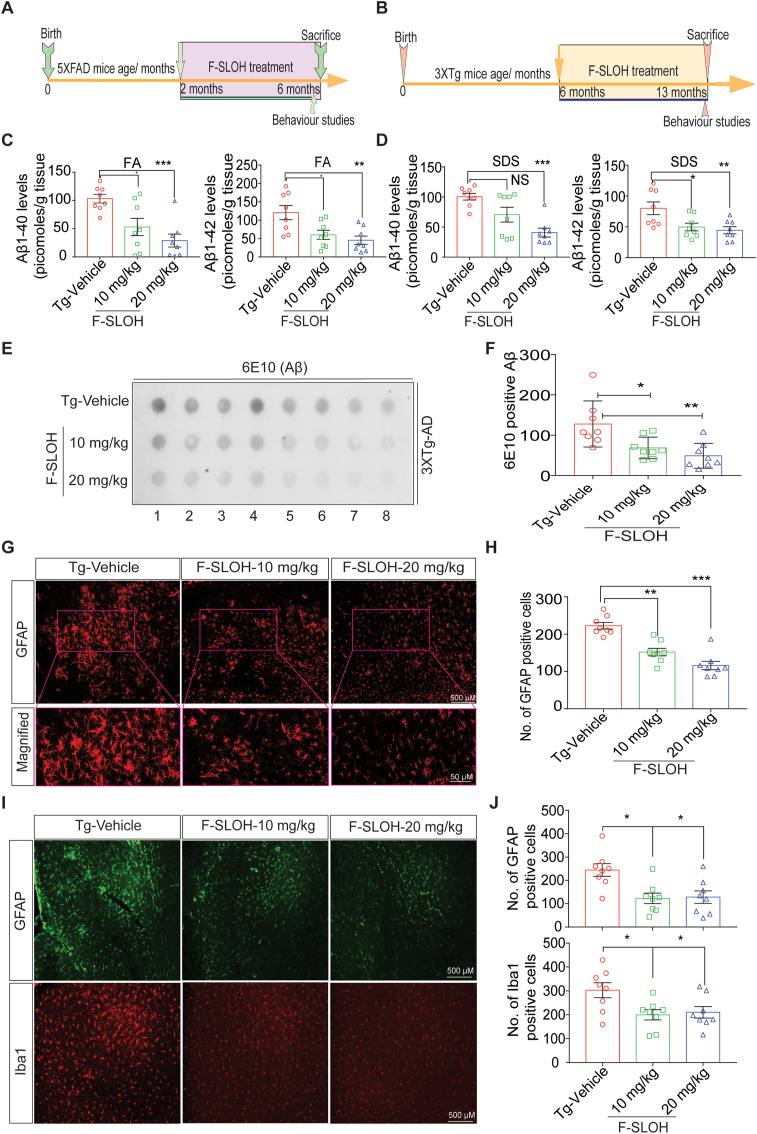
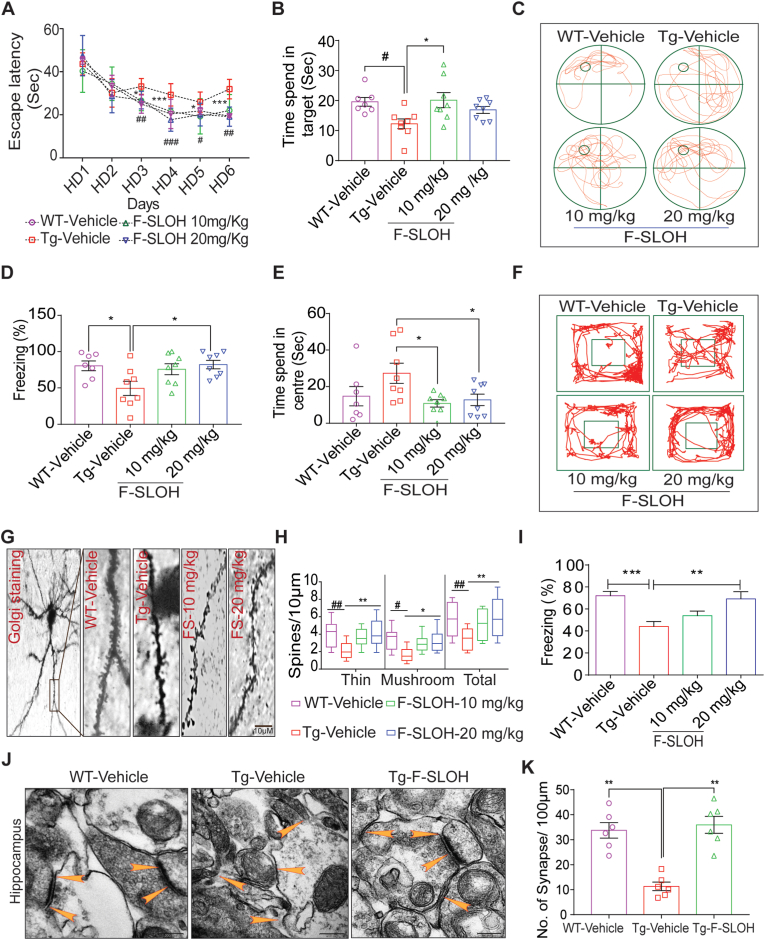
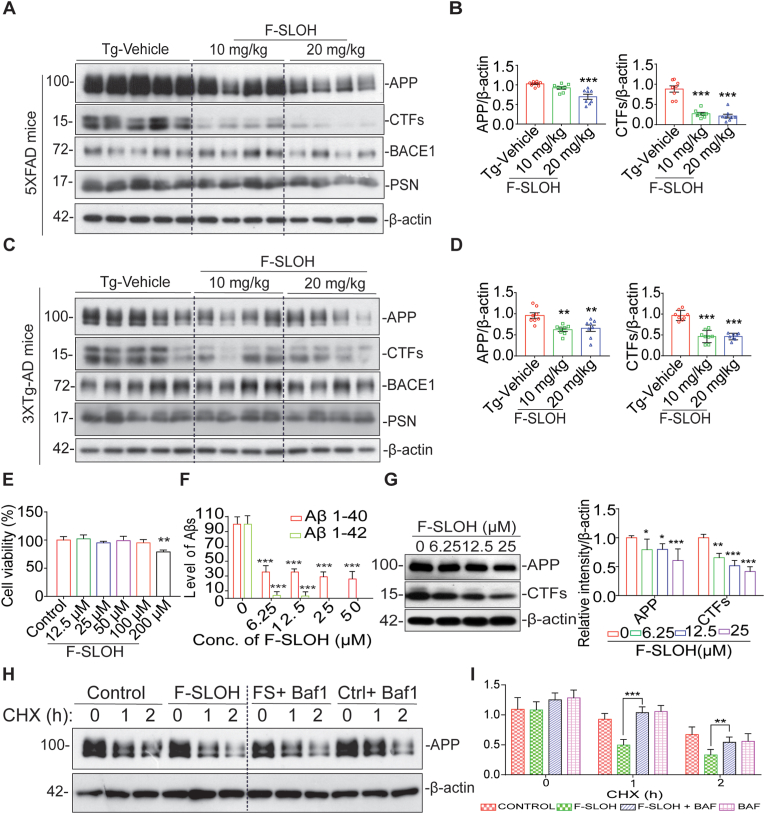
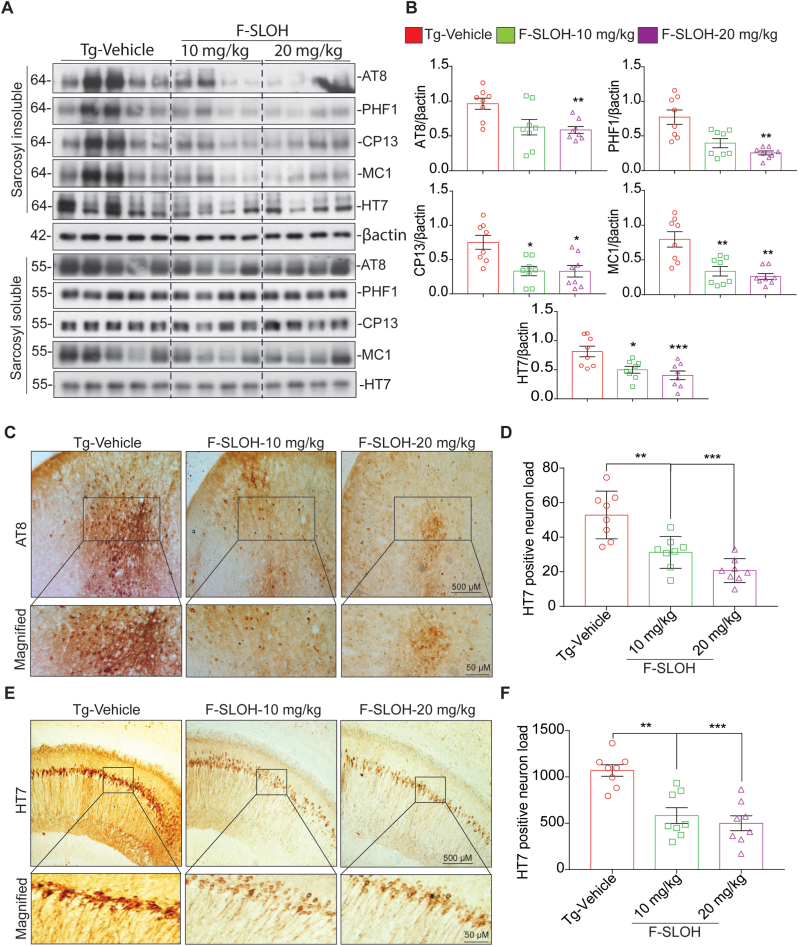
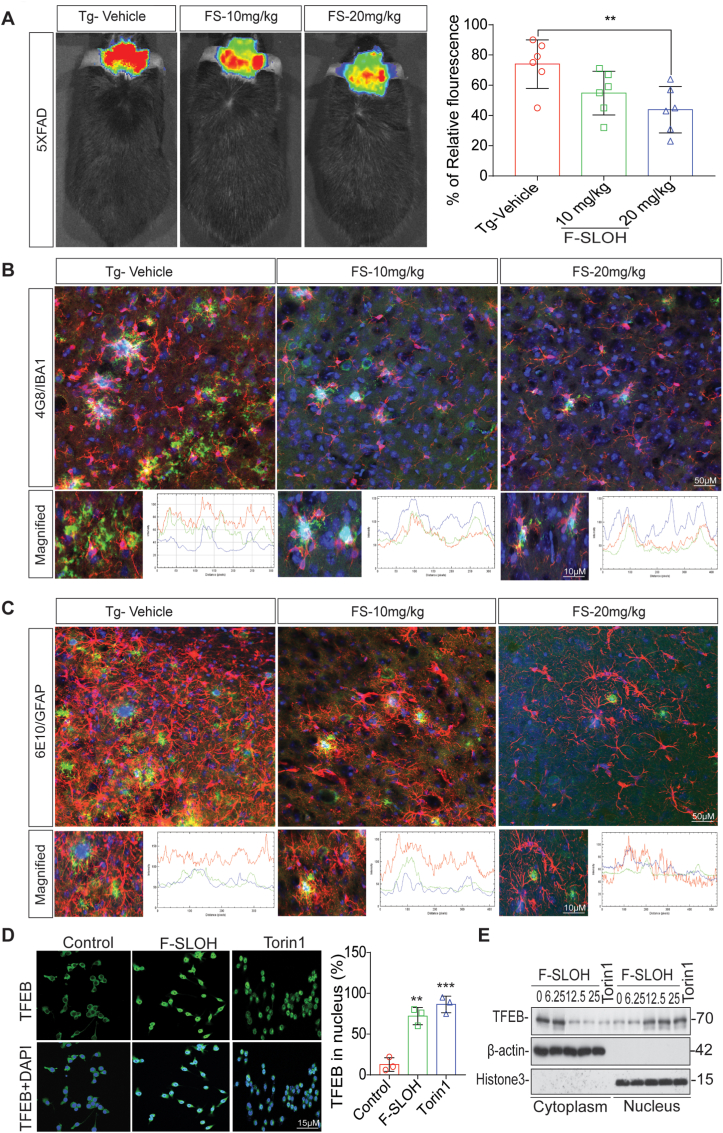
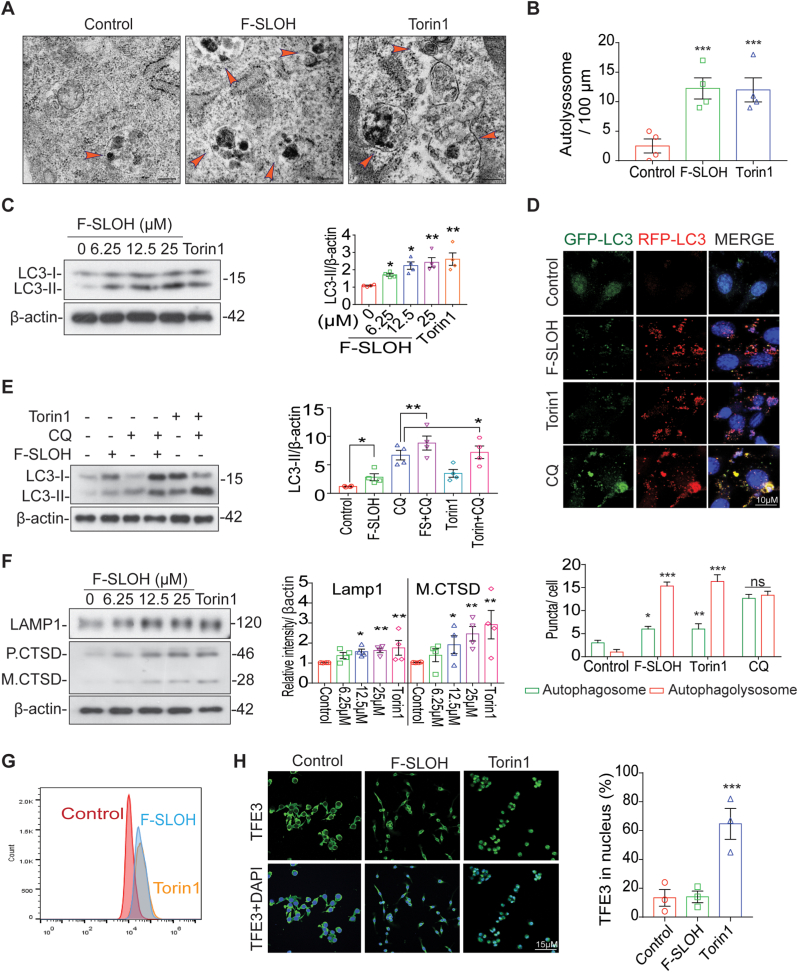
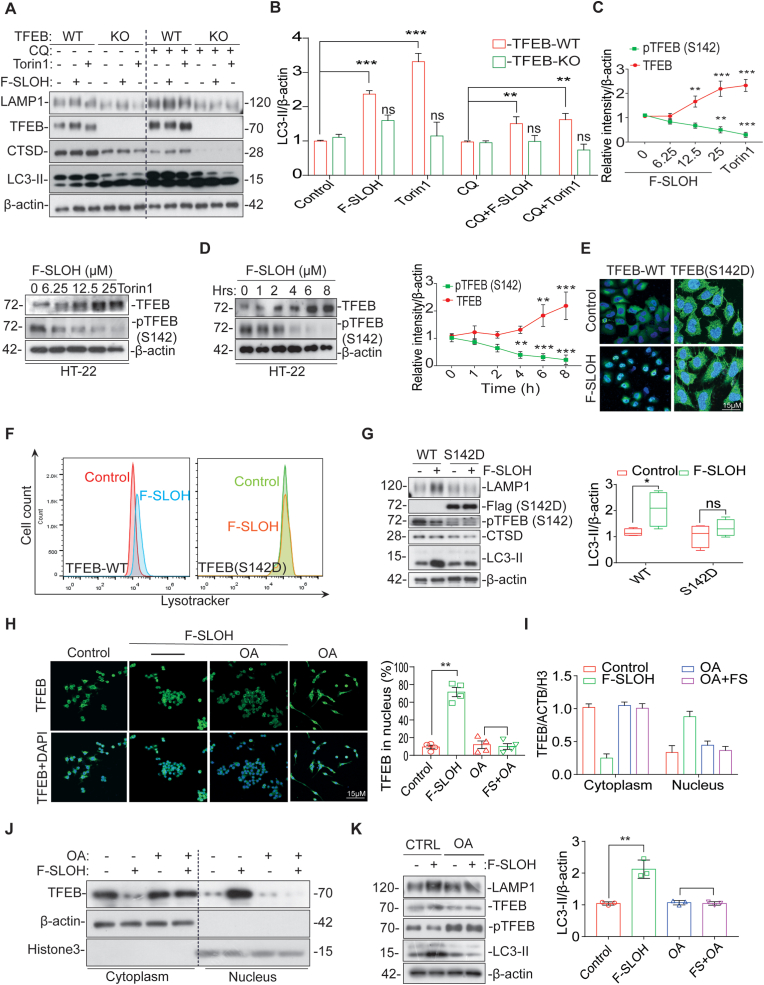
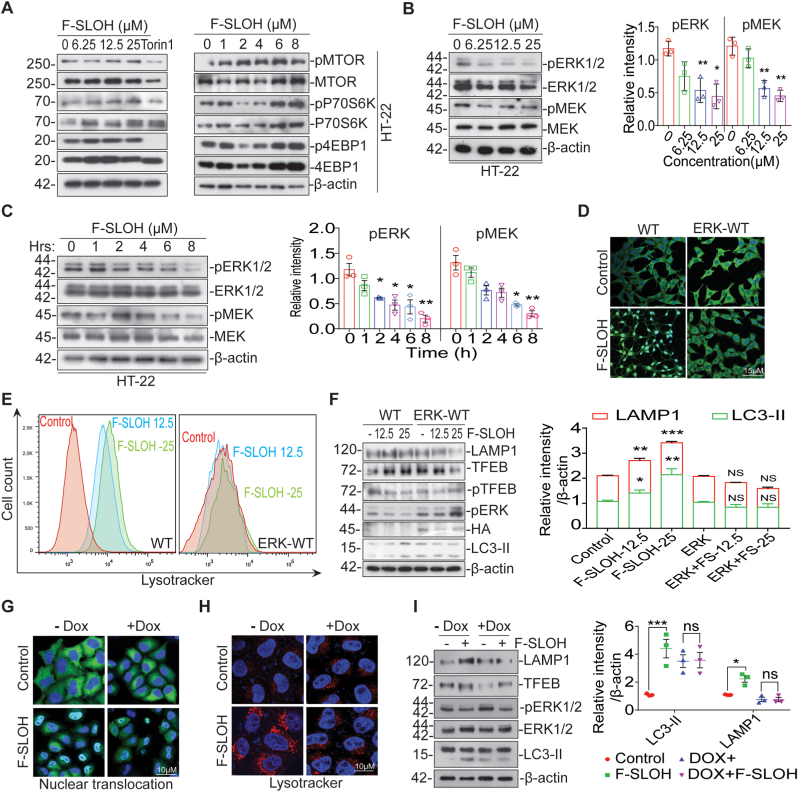
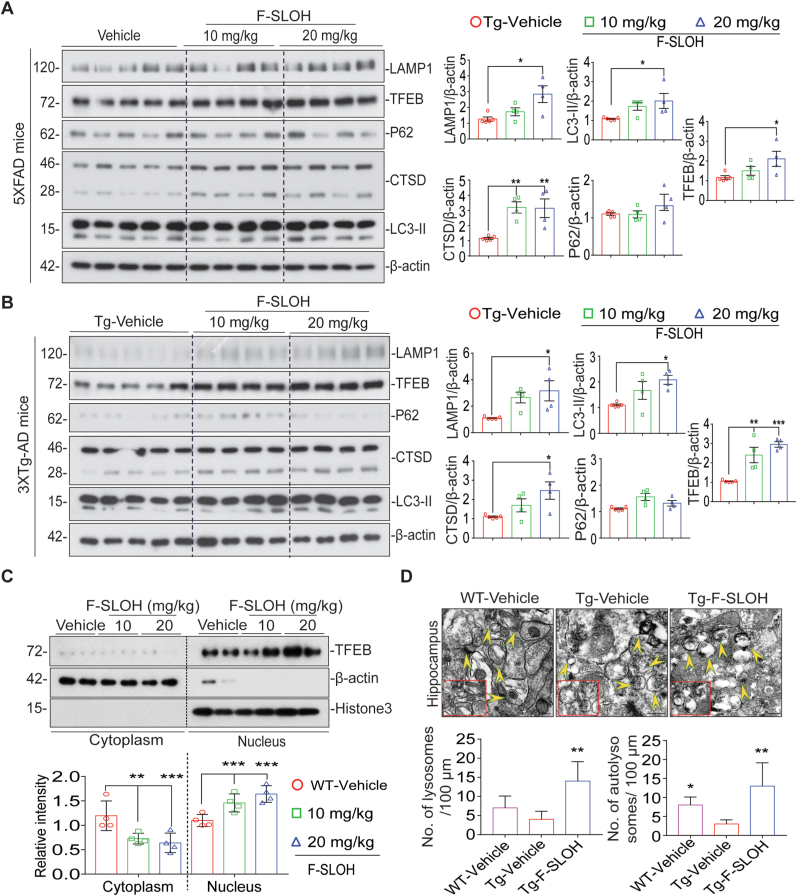
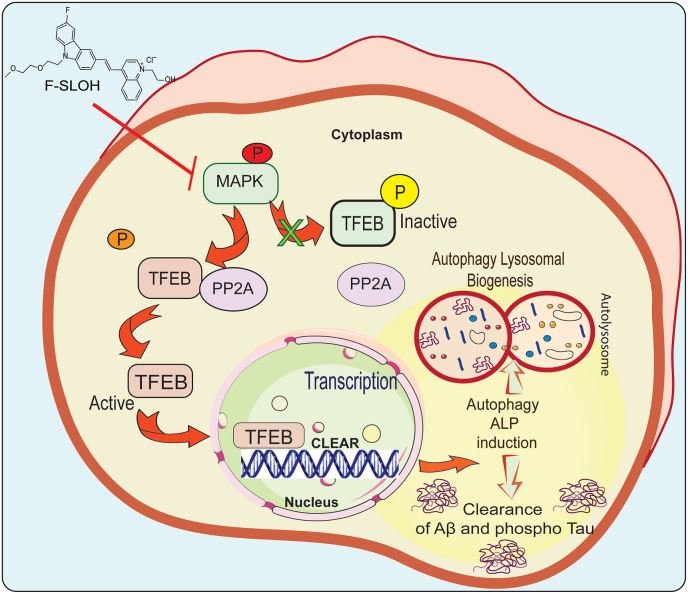
Accumulation of amyloid-β (Aβ) oligomers and phosphorylated Tau aggregates are crucial pathological events or factors that cause progressive neuronal loss, and cognitive impairments in Alzheimer's disease (AD). Current medications for AD have failed to halt, much less reverse this neurodegenerative disorder; therefore, there is an urgent need for the development of effective and safe drugs for AD therapy. In the present study, the in vivo therapeutic efficacy of an Aβ-oligomer-targeted fluorescent probe, F-SLOH, was extensively investigated in 5XFAD and 3XTg-AD mouse models. We have shown that F-SLOH exhibits an efficient inhibitory activity against Aβ aggregation in vivo, and acts as an effective theranostic agent for the treatment of multiple neuropathological changes in AD mouse models. F-SLOH has been found to significantly reduce not only the levels of Aβ oligomers, Tau aggregates and plaques but also the levels of amyloid precursor protein (APP) and its metabolites via autophagy lysosomal degradation pathway (ALP) in the brains of 5XFAD and 3XTg-AD mice. It also reduces astrocyte activation and microgliosis ultimately alleviating neuro-inflammation. Furthermore, F-SLOH mitigates hyperphosphorylated Tau aggregates, synaptic deficits and ameliorates synaptic memory function, and cognitive impairment in AD mouse models. The mechanistic studies have shown that F-SLOH promotes the clearance of C-terminal fragment 15 (CTF15) of APP and Paired helical filaments of Tau (PHF1) in stable cell models via the activation of transcription factor EB (TFEB). Moreover, F-SLOH promotes ALP and lysosomal biogenesis for the clearance of soluble, insoluble Aβ, and phospho Tau. Our results unambiguously reveal effective etiological capabilities of theranostic F-SLOH to target and intervene multiple neuropathological changes in AD mouse models. Therefore, F-SLOH demonstrates tremendous therapeutic potential for treating AD in its early stage.
Keywords: 3XTg-AD; 5XFAD; Alzheimer's disease; Aβ-aggregate inhibition; Aβ-targeting; Theranostic.
Copyright © 2022 The Authors. Published by Elsevier B.V. All rights reserved.
Conflict of interest statement
The authors declare that they do not have any known competing financial interests and that personal relationships did not influence the research work reported in this manuscript.
Figures
References
-
- Hardy J. A hundred years of Alzheimer's disease research. Neuron. 2006;52:3–13. - PubMed
-
- Alzheimer's A. 2016 Alzheimer's disease facts and figures. Alzheimers Dement. 2016;12:459–509. - PubMed
-
- Brookmeyer R., Johnson E., Ziegler-Graham K., Arrighi H.M. Forecasting the global burden of Alzheimer's disease. Alzheimers Dement. 2007;3:186–191. - PubMed
-
- 2020 Alzheimer's disease facts and figures. Alzheimers Dement. 2020 - PubMed
-
- Sengoku R. Aging and Alzheimer's disease pathology. Neuropathology. 2020;40:22–29. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Research Materials
