

Establishing analytical validity of BeadChip array genotype data by comparison to whole-genome sequence and standard benchmark datasets
- PMID: 35287663
- PMCID: PMC8919546
- DOI: 10.1186/s12920-022-01199-8
Establishing analytical validity of BeadChip array genotype data by comparison to whole-genome sequence and standard benchmark datasets
Abstract
Background: Clinical use of genotype data requires high positive predictive value (PPV) and thorough understanding of the genotyping platform characteristics. BeadChip arrays, such as the Global Screening Array (GSA), potentially offer a high-throughput, low-cost clinical screen for known variants. We hypothesize that quality assessment and comparison to whole-genome sequence and benchmark data establish the analytical validity of GSA genotyping.
Methods: To test this hypothesis, we selected 263 samples from Coriell, generated GSA genotypes in triplicate, generated whole genome sequence (rWGS) genotypes, assessed the quality of each set of genotypes, and compared each set of genotypes to each other and to the 1000 Genomes Phase 3 (1KG) genotypes, a performance benchmark. For 59 genes (MAP59), we also performed theoretical and empirical evaluation of variants deemed medically actionable predispositions.
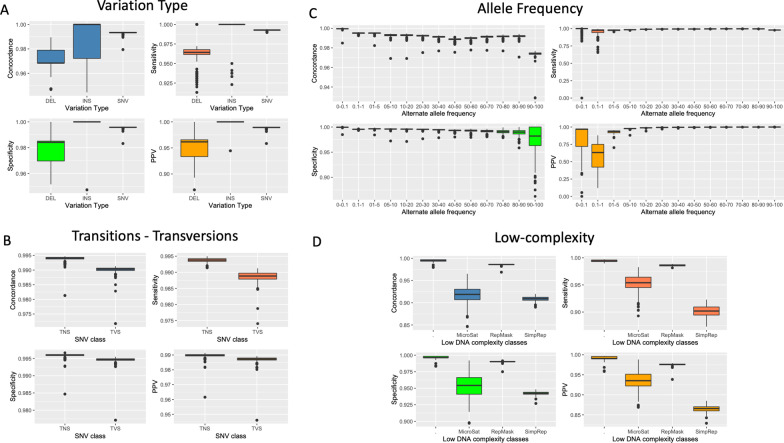
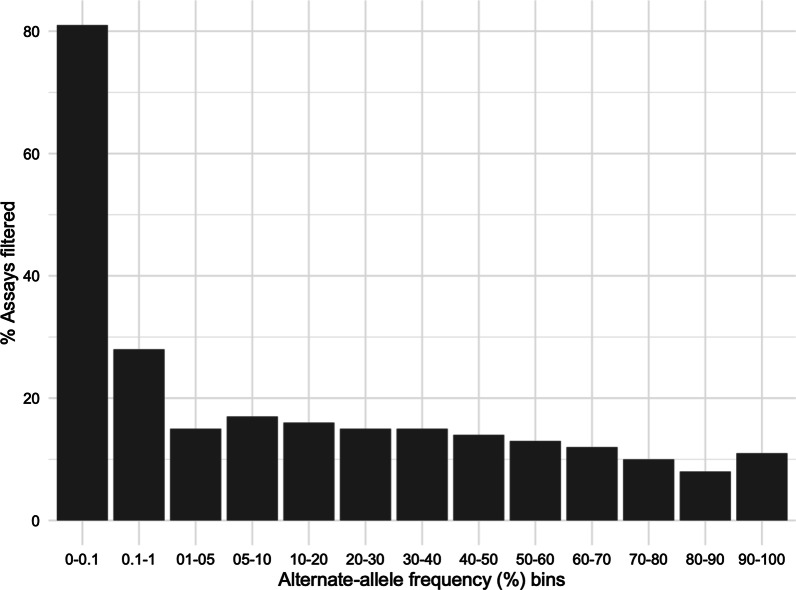
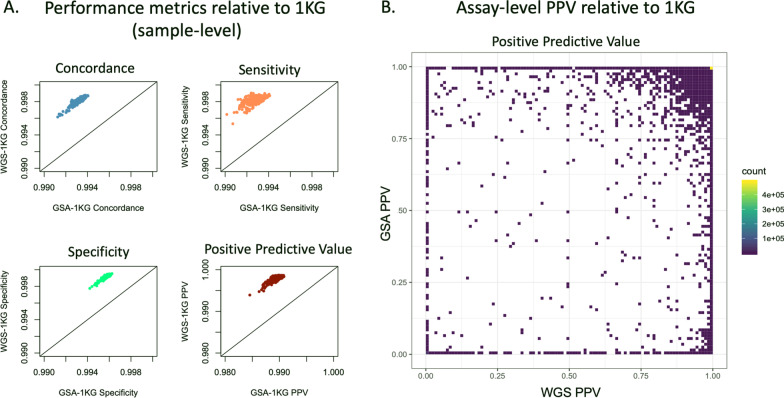
Results: Quality analyses detected sample contamination and increased assay failure along the chip margins. Comparison to benchmark data demonstrated that > 82% of the GSA assays had a PPV of 1. GSA assays targeting transitions, genomic regions of high complexity, and common variants performed better than those targeting transversions, regions of low complexity, and rare variants. Comparison of GSA data to rWGS and 1KG data showed > 99% performance across all measured parameters. Consistent with predictions from prior studies, the GSA detection of variation within the MAP59 genes was 3/261.
Conclusion: We establish the analytical validity of GSA assays using quality analytics and comparison to benchmark and rWGS data. GSA assays meet the standards of a clinical screen although assays interrogating rare variants, transversions, and variants within low-complexity regions require careful evaluation.
Keywords: Analytical validation; Clinical genotyping; Genotyping error.
© 2022. The Author(s).
Conflict of interest statement
The authors declare that they have no competing interests.
Figures




References
-
- Walters K. The effect of genotyping error in sib-pair genomewide linkage scans depends crucially upon the method of analysis. J Hum Genet. 2005;50(7):329–337. - PubMed
-
- Saunders IW, Brohede J, Hannan GN. Estimating genotyping error rates from Mendelian errors in SNP array genotypes and their impact on inference. Genomics. 2007;90(3):291–296. - PubMed
-
- Pompanon F, Bonin A, Bellemain E, Taberlet P. Genotyping errors: causes, consequences and solutions. Nat Rev Genet. 2005;6(11):847–859. - PubMed
MeSH terms
LinkOut - more resources
Full Text Sources
