

Cell type-specific mechanistic target of rapamycin-dependent distortion of autophagy pathways in lupus nephritis
- PMID: 35288362
- PMCID: PMC9240418
- DOI: 10.1016/j.trsl.2022.03.004
Cell type-specific mechanistic target of rapamycin-dependent distortion of autophagy pathways in lupus nephritis
Abstract
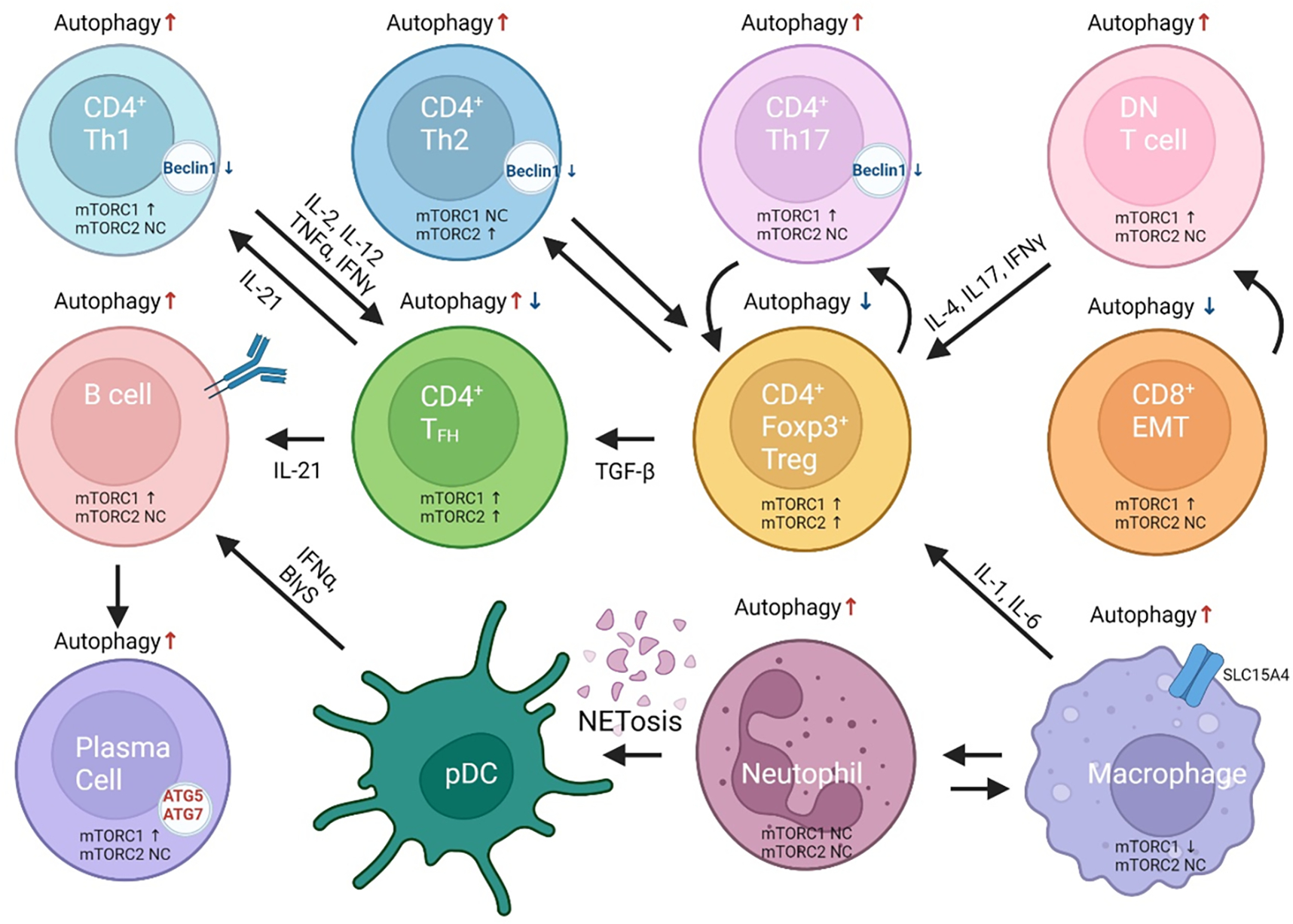
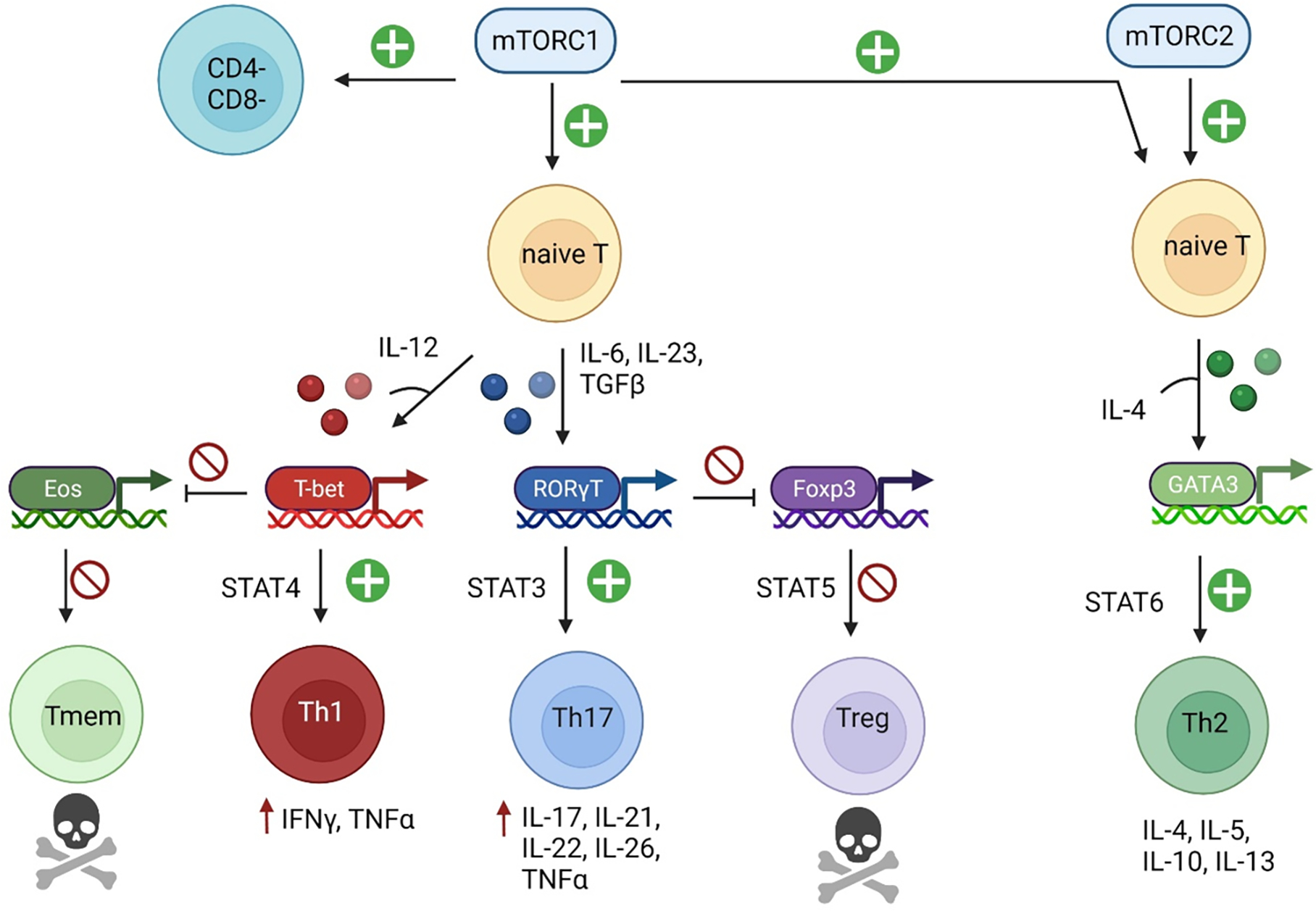
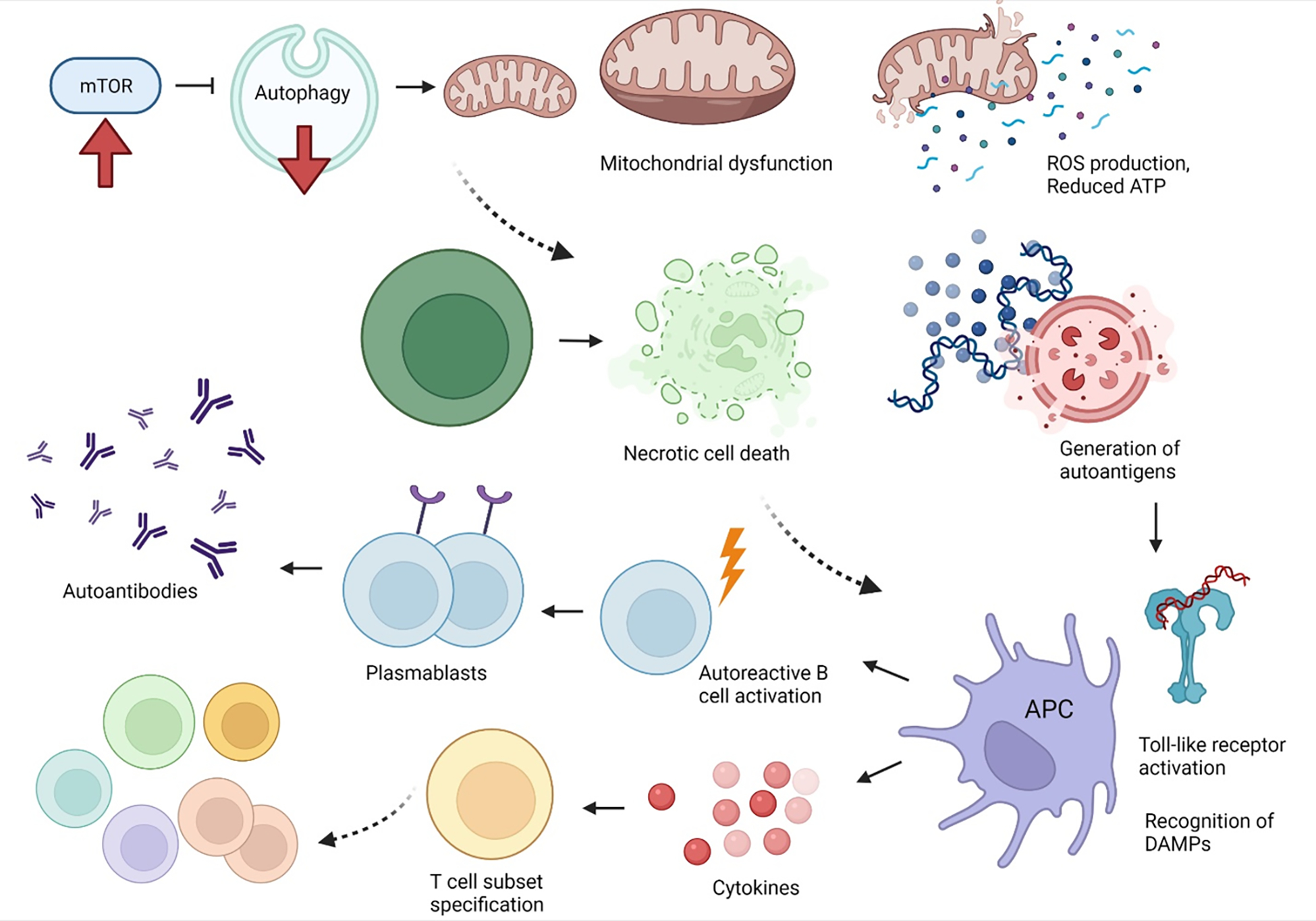
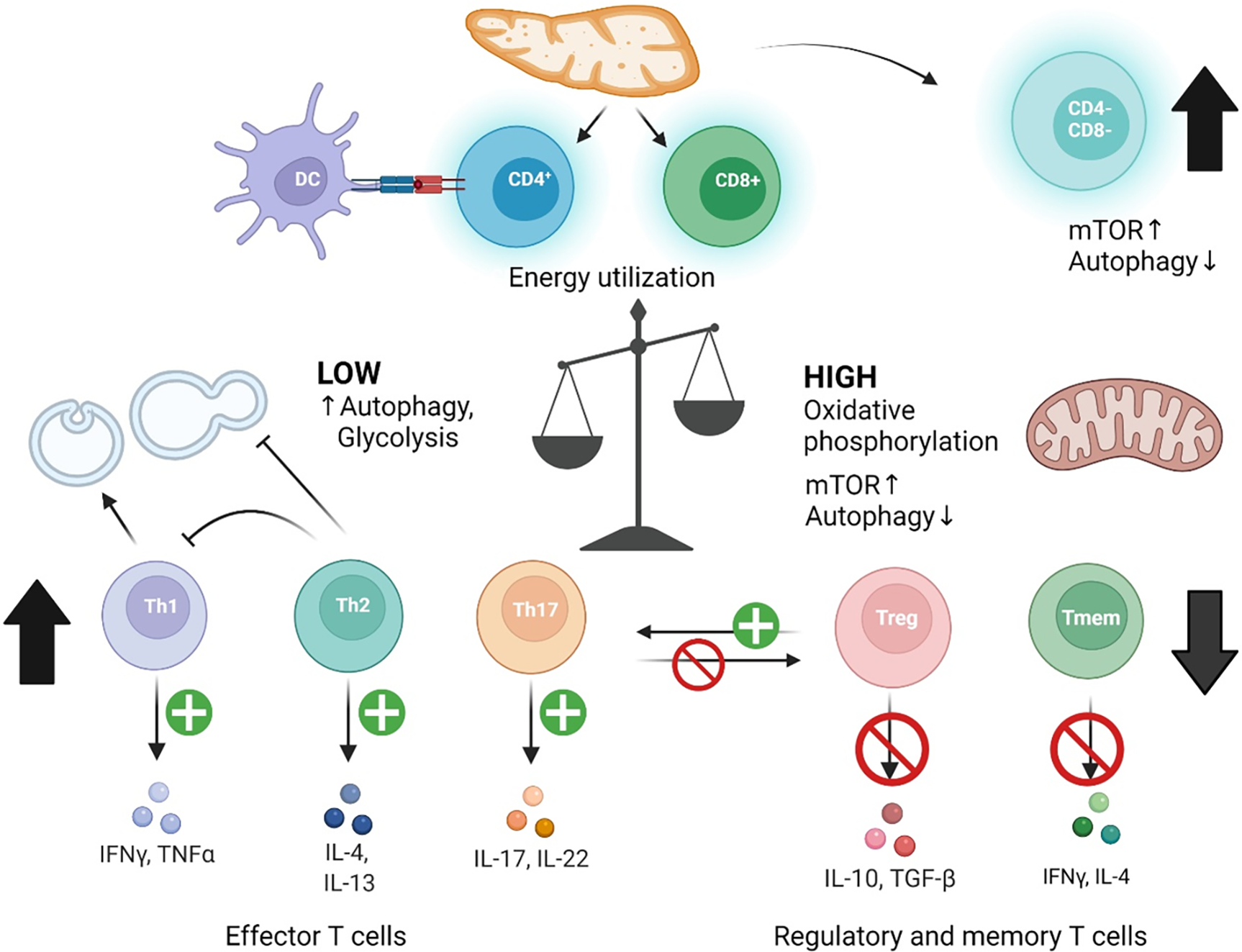
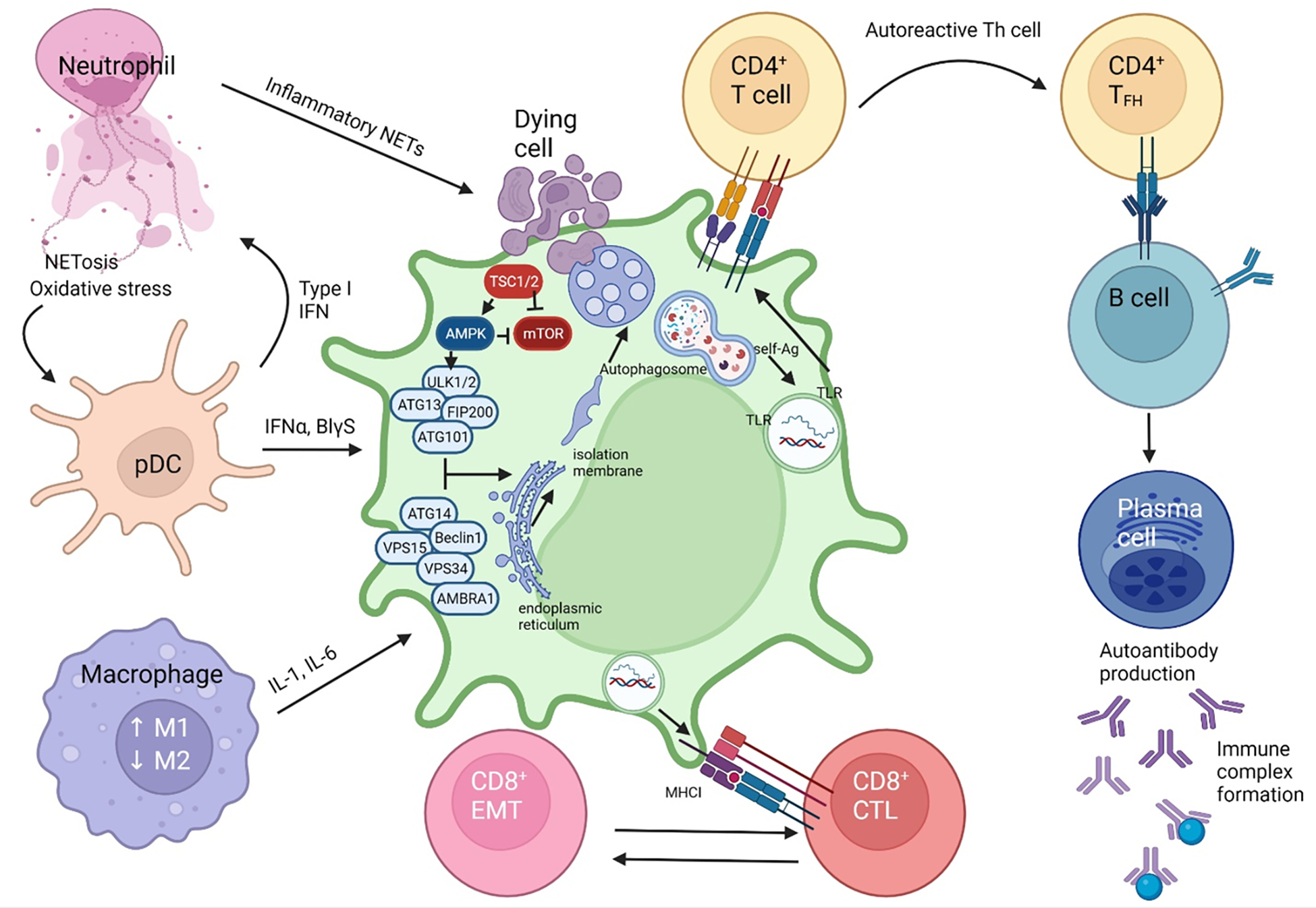
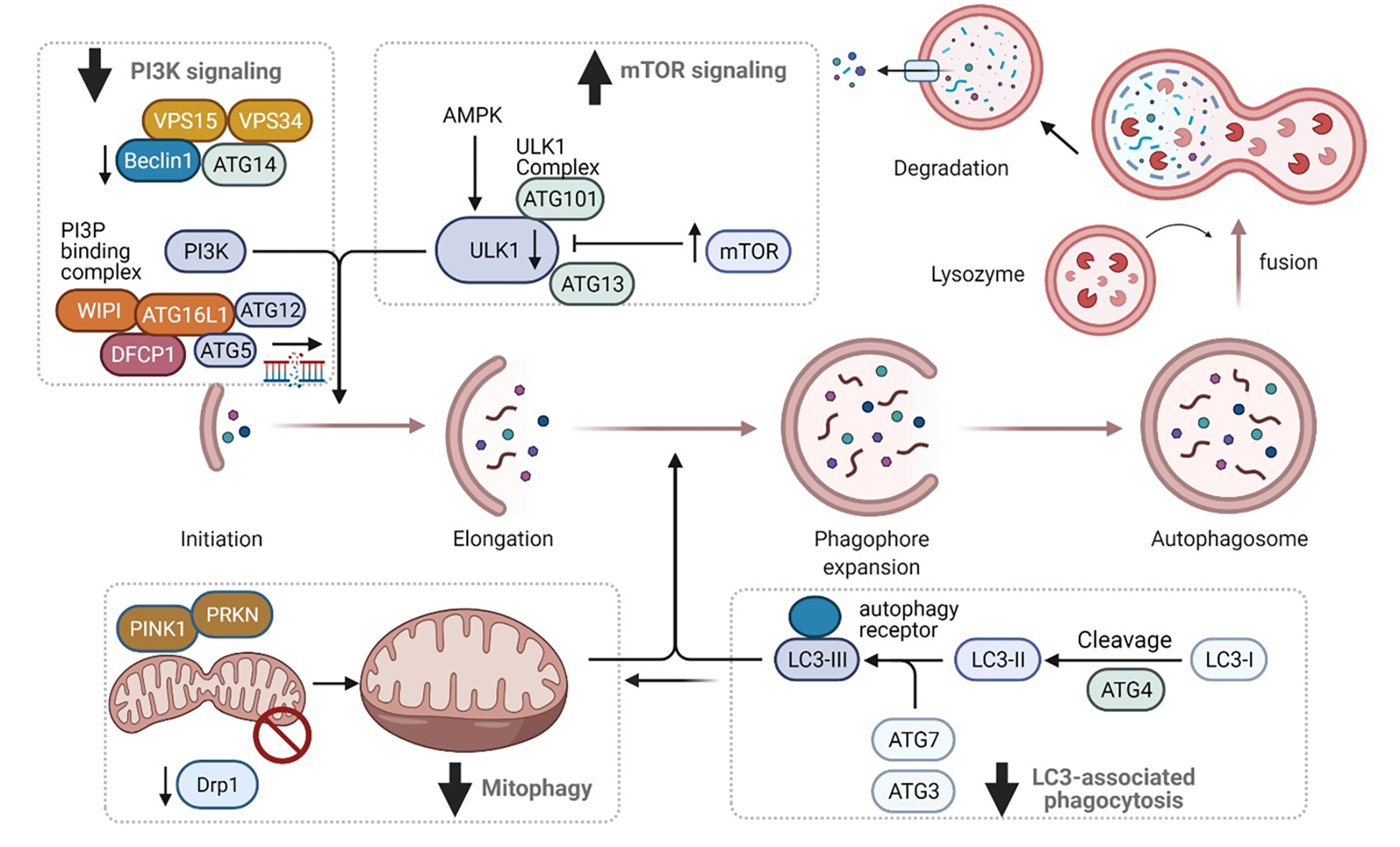
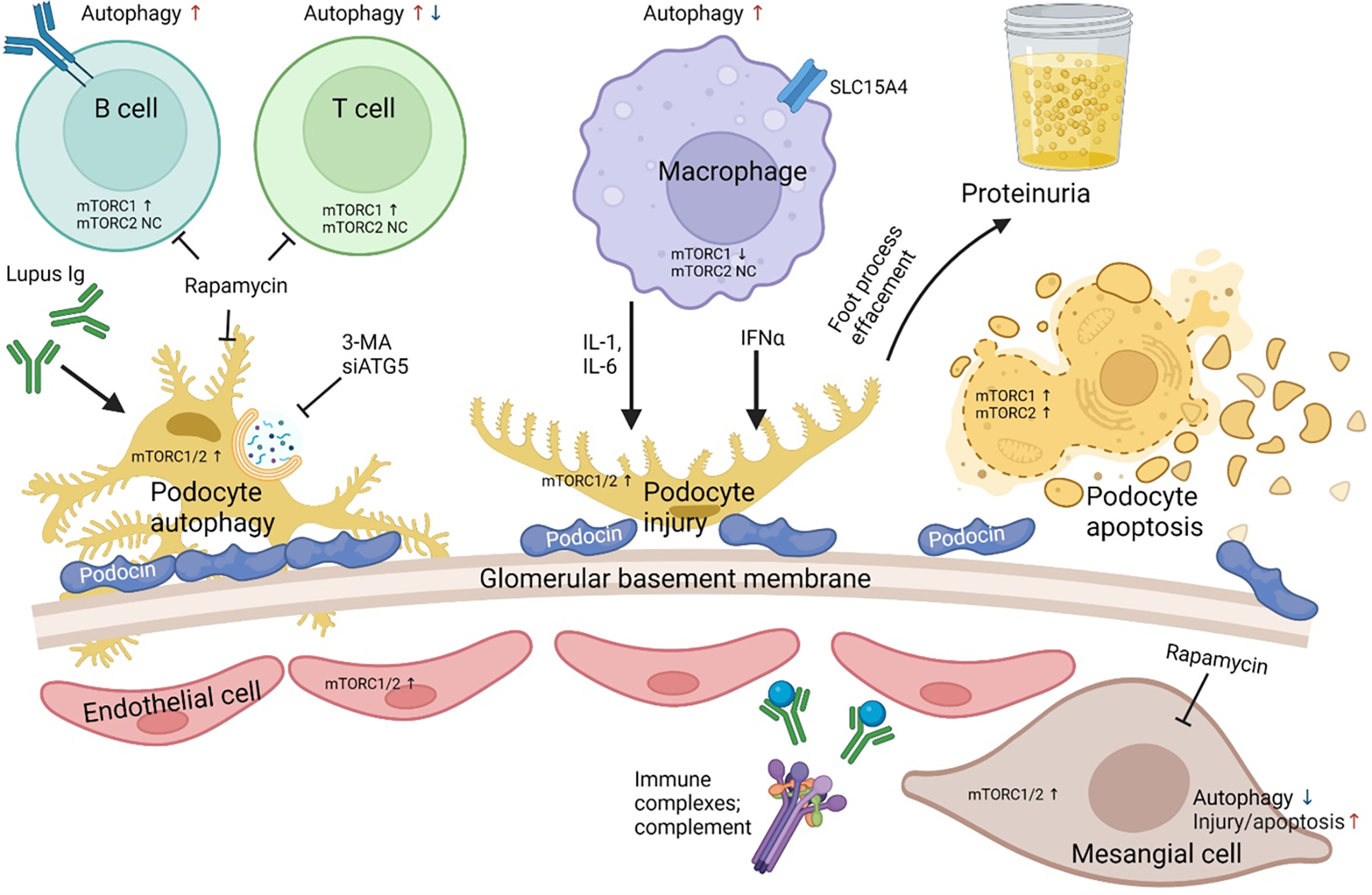
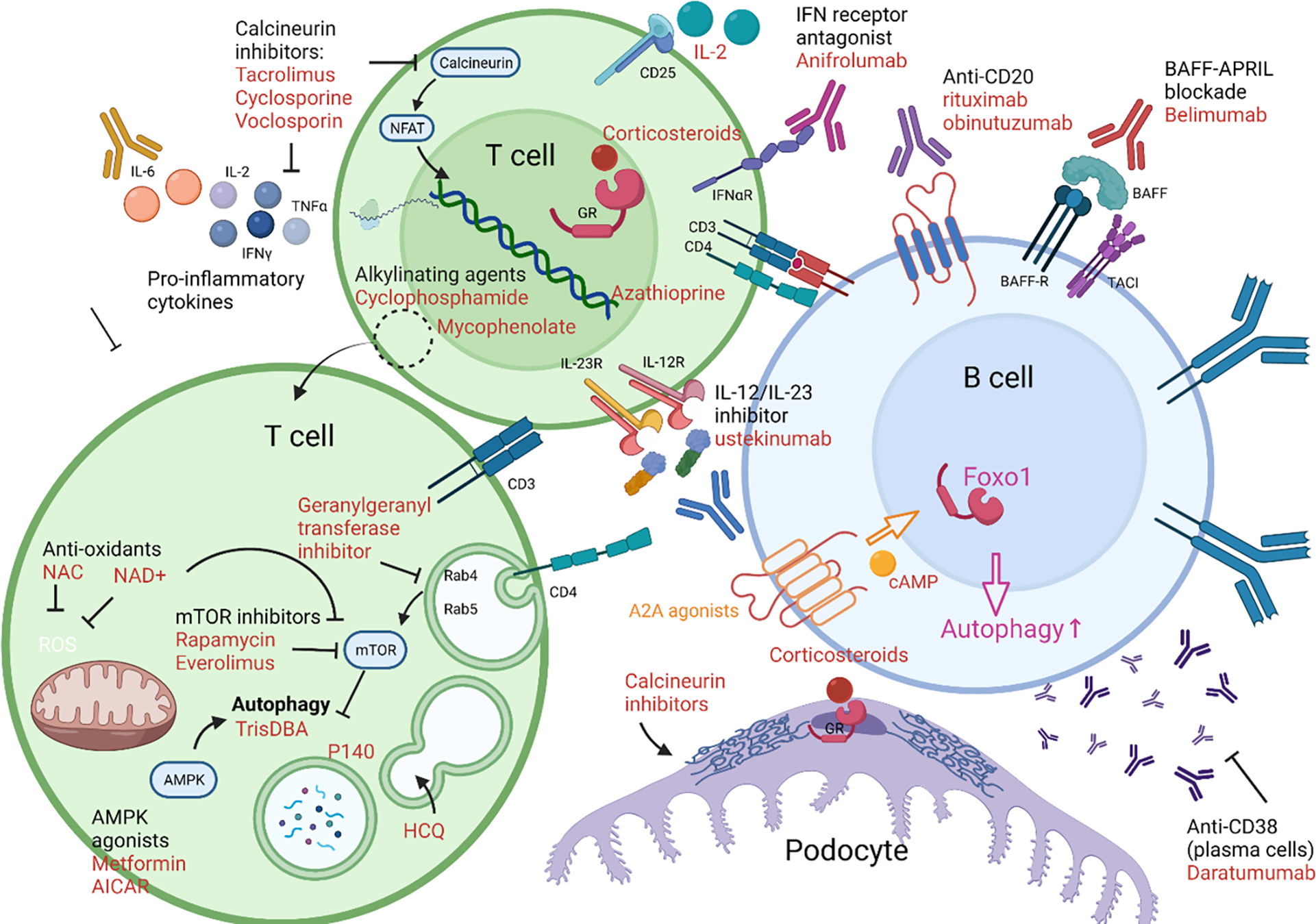
Pro-inflammatory immune system development, metabolomic defects, and deregulation of autophagy play interconnected roles in driving the pathogenesis of systemic lupus erythematosus (SLE). Lupus nephritis (LN) is a leading cause of morbidity and mortality in SLE. While the causes of SLE have not been clearly delineated, skewing of T and B cell differentiation, activation of antigen-presenting cells, production of antinuclear autoantibodies and pro-inflammatory cytokines are known to contribute to disease development. Underlying this process are defects in autophagy and mitophagy that cause the accumulation of oxidative stress-generating mitochondria which promote necrotic cell death. Autophagy is generally inhibited by the activation of the mammalian target of rapamycin (mTOR), a large protein kinase that underlies abnormal immune cell lineage specification in SLE. Importantly, several autophagy-regulating genes, including ATG5 and ATG7, as well as mitophagy-regulating HRES-1/Rab4A have been linked to lupus susceptibility and molecular pathogenesis. Moreover, genetically-driven mTOR activation has been associated with fulminant lupus nephritis. mTOR activation and diminished autophagy promote the expansion of pro-inflammatory Th17, Tfh and CD3+CD4-CD8- double-negative (DN) T cells at the expense of CD8+ effector memory T cells and CD4+ regulatory T cells (Tregs). mTOR activation and aberrant autophagy also involve renal podocytes, mesangial cells, endothelial cells, and tubular epithelial cells that may compromise end-organ resistance in LN. Activation of mTOR complexes 1 (mTORC1) and 2 (mTORC2) has been identified as biomarkers of disease activation and predictors of disease flares and prognosis in SLE patients with and without LN. This review highlights recent advances in molecular pathogenesis of LN with a focus on immuno-metabolic checkpoints of autophagy and their roles in pathogenesis, prognosis and selection of targets for treatment in SLE.
Copyright © 2022 Elsevier Inc. All rights reserved.
Conflict of interest statement
The authors have read the journal’s policy on disclosure of potential conflicts of interest and declare no conflicts of interest. All authors have read the journal’s authorship agreement and that the manuscript has been reviewed by and approved by all named authors.
Figures
Similar articles
-
Blockade of Treg Cell Differentiation and Function by the Interleukin-21-Mechanistic Target of Rapamycin Axis Via Suppression of Autophagy in Patients With Systemic Lupus Erythematosus.Arthritis Rheumatol. 2018 Mar;70(3):427-438. doi: 10.1002/art.40380. Epub 2018 Jan 30. Arthritis Rheumatol. 2018. PMID: 29161463 Free PMC article.
-
Mechanistic target of rapamycin complex 1 expands Th17 and IL-4+ CD4-CD8- double-negative T cells and contracts regulatory T cells in systemic lupus erythematosus.J Immunol. 2014 May 1;192(9):4134-44. doi: 10.4049/jimmunol.1301859. Epub 2014 Mar 28. J Immunol. 2014. PMID: 24683191 Free PMC article.
-
Rab4A-directed endosome traffic shapes pro-inflammatory mitochondrial metabolism in T cells via mitophagy, CD98 expression, and kynurenine-sensitive mTOR activation.Nat Commun. 2024 Mar 22;15(1):2598. doi: 10.1038/s41467-024-46441-2. Nat Commun. 2024. PMID: 38519468 Free PMC article.
-
Mitochondrial Dysfunction in Systemic Lupus Erythematosus with a Focus on Lupus Nephritis.Int J Mol Sci. 2024 Jun 3;25(11):6162. doi: 10.3390/ijms25116162. Int J Mol Sci. 2024. PMID: 38892349 Free PMC article. Review.
-
Calcineurin and Systemic Lupus Erythematosus: The Rationale for Using Calcineurin Inhibitors in the Treatment of Lupus Nephritis.Int J Mol Sci. 2021 Jan 27;22(3):1263. doi: 10.3390/ijms22031263. Int J Mol Sci. 2021. PMID: 33514066 Free PMC article. Review.
Cited by
-
Integrating genomics and AI to uncover molecular targets for mRNA vaccine development in lupus nephritis.Front Immunol. 2024 Oct 4;15:1381445. doi: 10.3389/fimmu.2024.1381445. eCollection 2024. Front Immunol. 2024. PMID: 39430760 Free PMC article.
-
Glomerular mTORC1 activation was associated with podocytes to endothelial cells communication in lupus nephritis.Lupus Sci Med. 2023 May;10(1):e000896. doi: 10.1136/lupus-2023-000896. Lupus Sci Med. 2023. PMID: 37147021 Free PMC article.
-
Sirolimus versus mycophenolate mofetil for the treatment of lupus nephritis: Results from a real-world CSTAR cohort study.Rheumatol Immunol Res. 2025 Jul 1;6(2):80-89. doi: 10.1515/rir-2025-0011. eCollection 2025 Jun. Rheumatol Immunol Res. 2025. PMID: 40606852 Free PMC article.
-
Immunometabolic alterations in lupus: where do they come from and where do we go from there?Curr Opin Immunol. 2022 Oct;78:102245. doi: 10.1016/j.coi.2022.102245. Epub 2022 Sep 16. Curr Opin Immunol. 2022. PMID: 36122544 Free PMC article. Review.
-
Rapamycin relieves lupus nephritis by regulating TIM-3 and CD4+CD25+Foxp3+ Treg cells in an MRL/lpr mouse model.Cent Eur J Immunol. 2022;47(3):206-217. doi: 10.5114/ceji.2022.118778. Epub 2022 Sep 15. Cent Eur J Immunol. 2022. PMID: 36817267 Free PMC article.
References
-
- Alarcón GS, Roseman J, Bartolucci AA, Friedman AW, Moulds JM, Goel N, et al. Systemic lupus erythematosus in three ethnic groups: II. Features predictive of disease activity early in its course. LUMINA Study Group. Lupus in minority populations, nature versus nurture. Arthritis Rheum. 1998;41(7):1173–80. Epub 1998/07/15. doi: 10.1002/1529-0131(199807)41:7<1173::Aid-art5>3.0.Co;2-a. - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
