

Population genomics of Escherichia coli in livestock-keeping households across a rapidly developing urban landscape
- PMID: 35288654
- PMCID: PMC8975746
- DOI: 10.1038/s41564-022-01079-y
Population genomics of Escherichia coli in livestock-keeping households across a rapidly developing urban landscape
Abstract
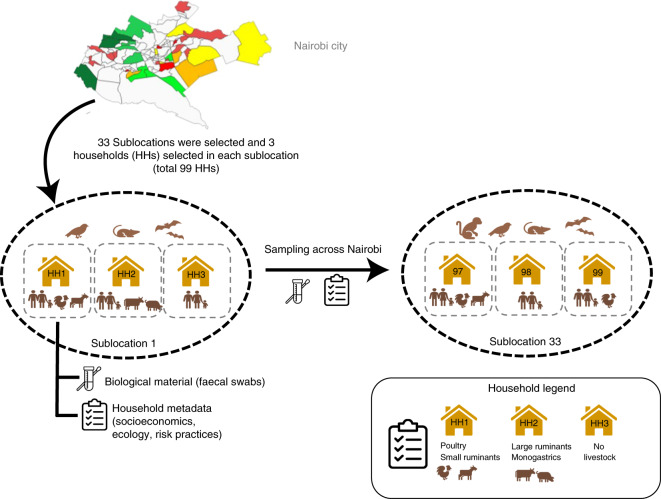
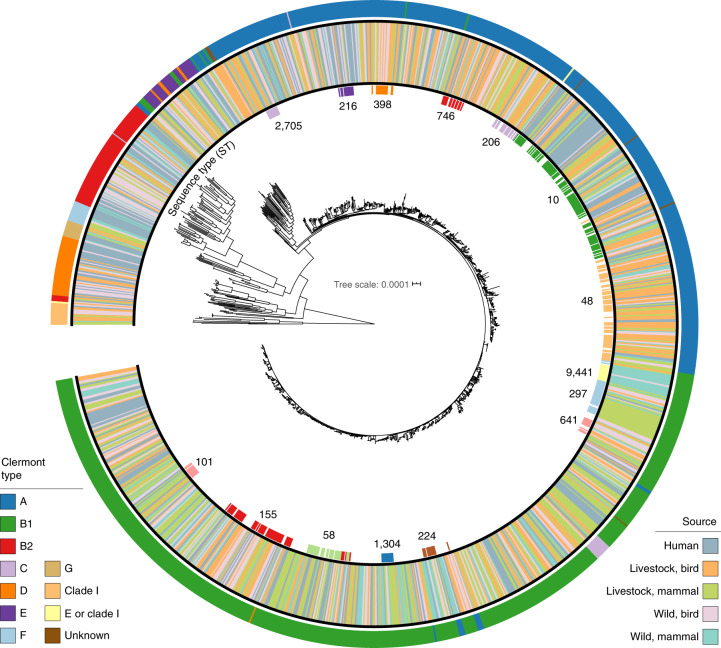
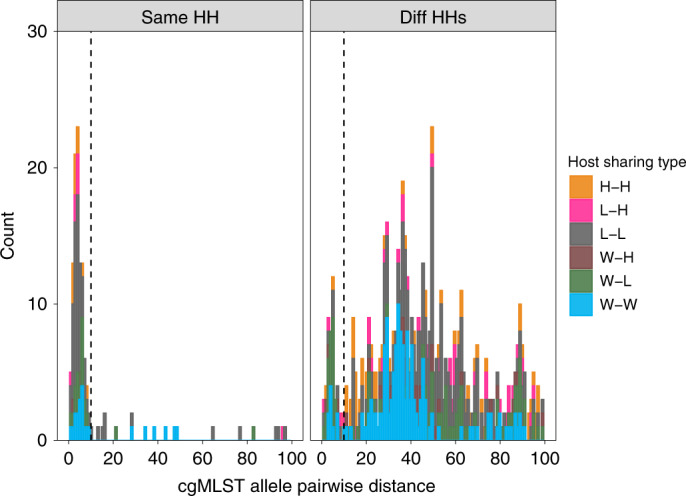
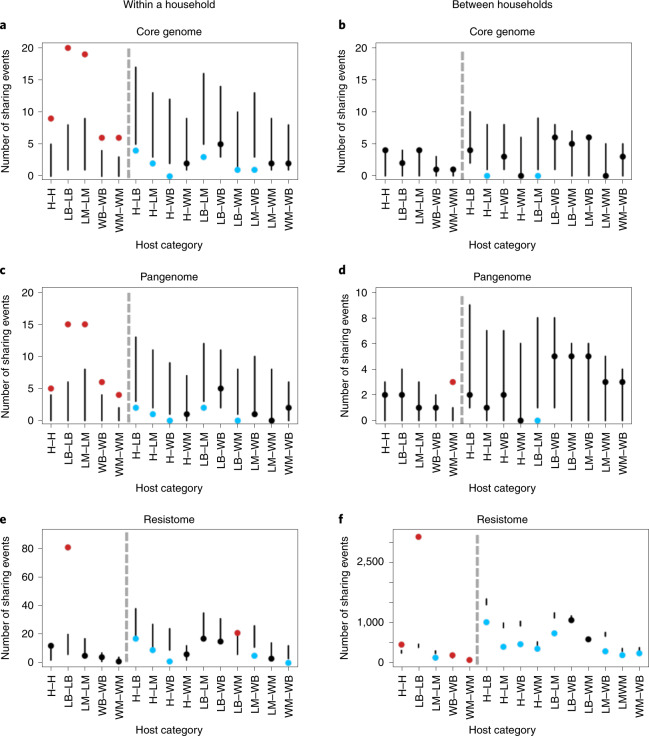
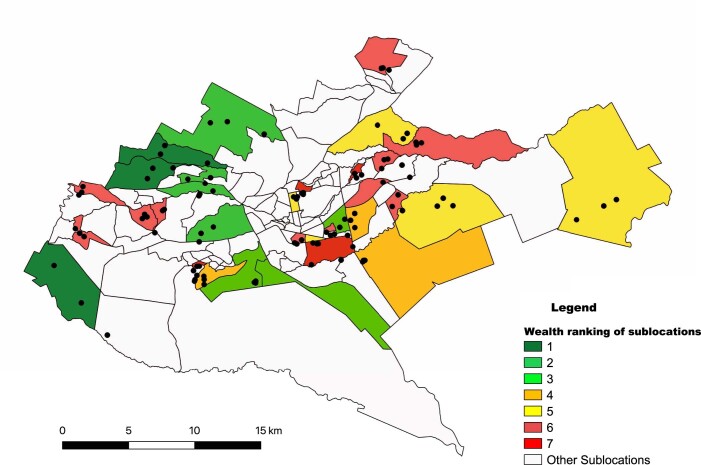
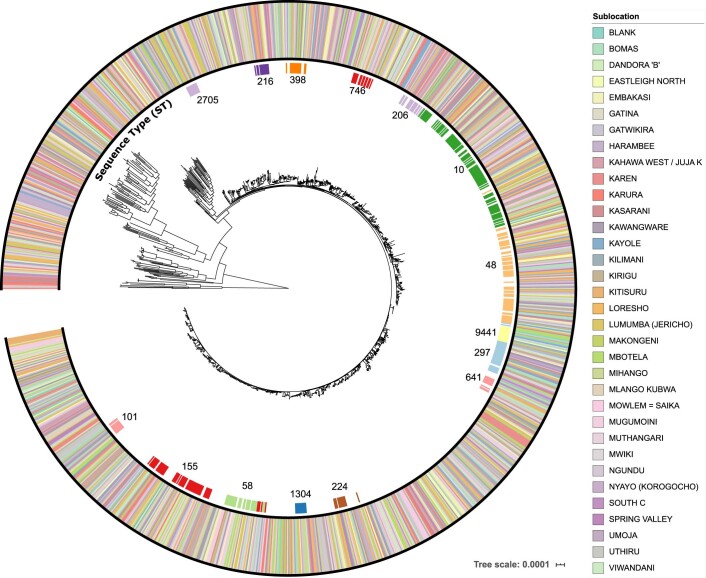
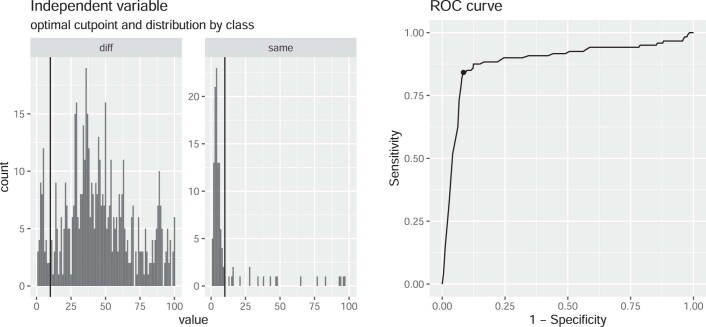
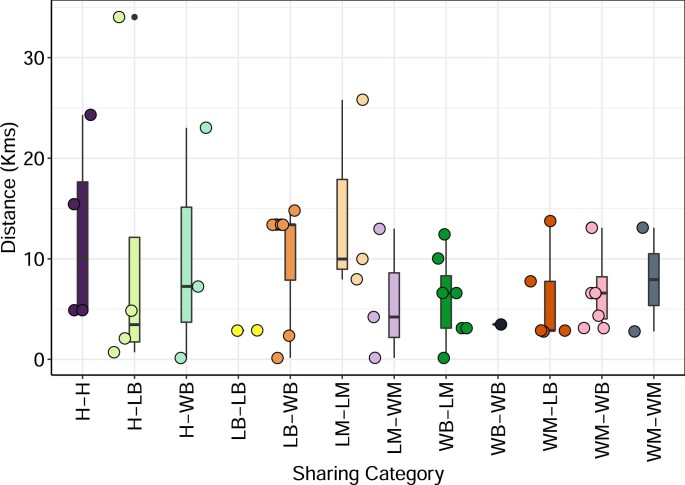
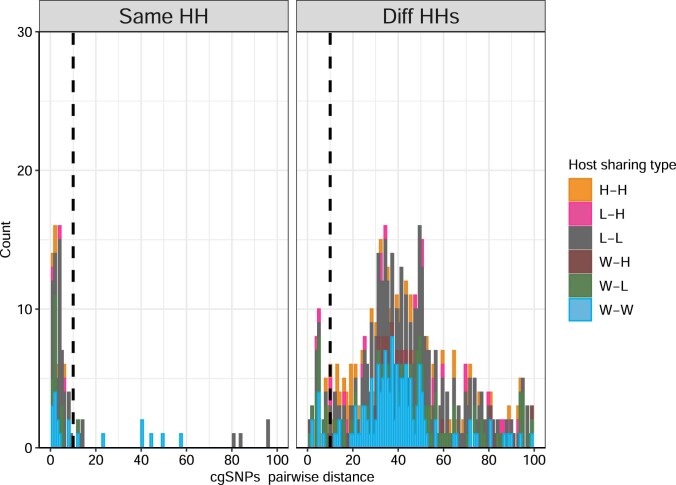
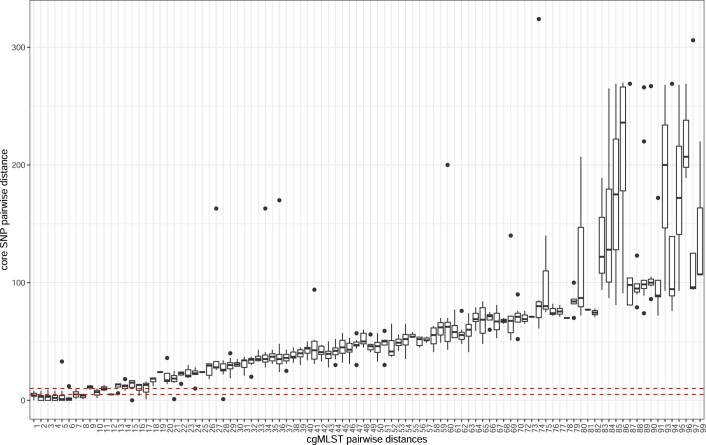
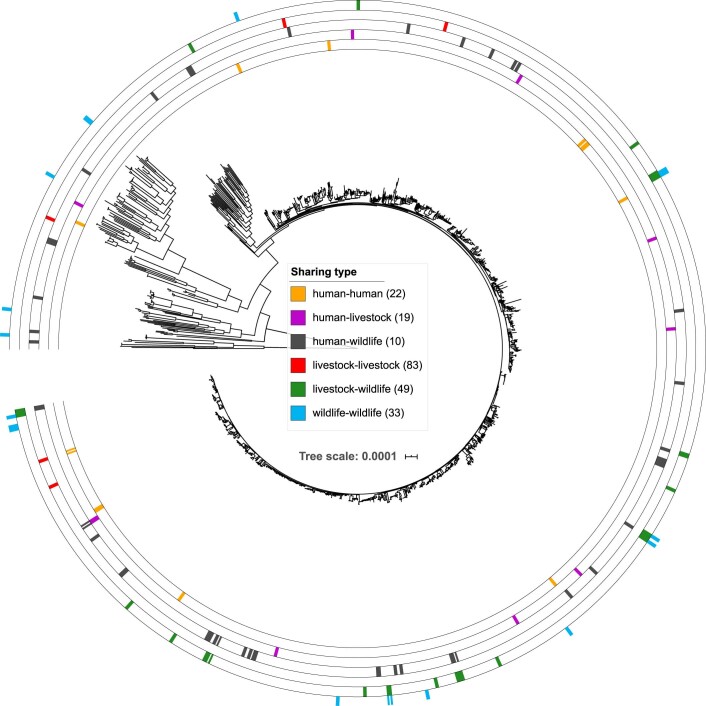
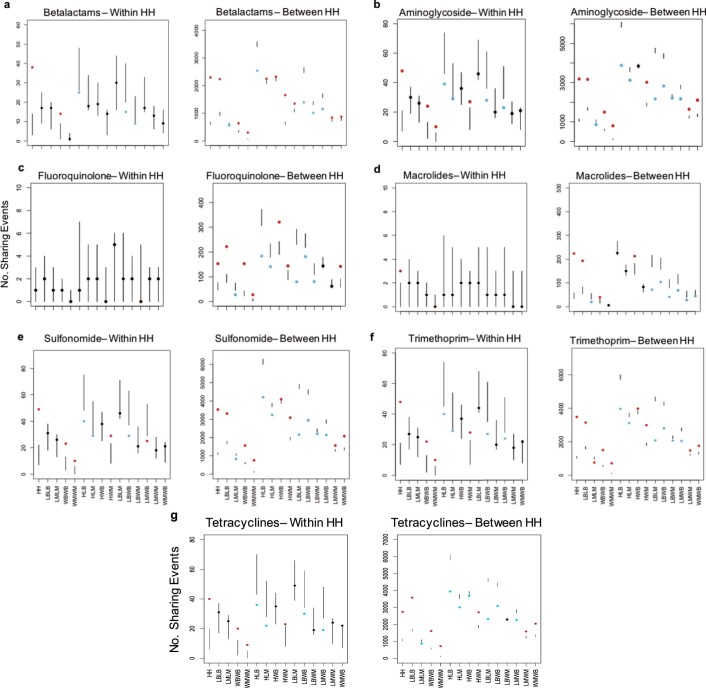
Quantitative evidence for the risk of zoonoses and the spread of antimicrobial resistance remains lacking. Here, as part of the UrbanZoo project, we sampled Escherichia coli from humans, livestock and peri-domestic wildlife in 99 households across Nairobi, Kenya, to investigate its distribution among host species in this rapidly developing urban landscape. We performed whole-genome sequencing of 1,338 E. coli isolates and found that the diversity and sharing patterns of E. coli were heavily structured by household and strongly shaped by host type. We also found evidence for inter-household and inter-host sharing and, importantly, between humans and animals, although this occurs much less frequently. Resistome similarity was differently distributed across host and household, consistent with being driven by shared exposure to antimicrobials. Our results indicate that a large, epidemiologically structured sampling framework combined with WGS is needed to uncover strain-sharing events among different host populations in complex environments and the major contributing pathways that could ultimately drive the emergence of zoonoses and the spread of antimicrobial resistance.
© 2022. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
Comment in
-
Baas Becking meets One Health.Nat Microbiol. 2022 Apr;7(4):482-483. doi: 10.1038/s41564-022-01100-4. Nat Microbiol. 2022. PMID: 35365789 No abstract available.
References
-
- Gottdenker NL, Streicker DG, Faust CL, Carroll CR. Anthropogenic land use change and infectious diseases: a review of the evidence. EcoHealth. 2014;11:619–632. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
