

Systematic reconstruction of cellular trajectories across mouse embryogenesis
- PMID: 35288709
- PMCID: PMC8920898
- DOI: 10.1038/s41588-022-01018-x
Systematic reconstruction of cellular trajectories across mouse embryogenesis
Abstract
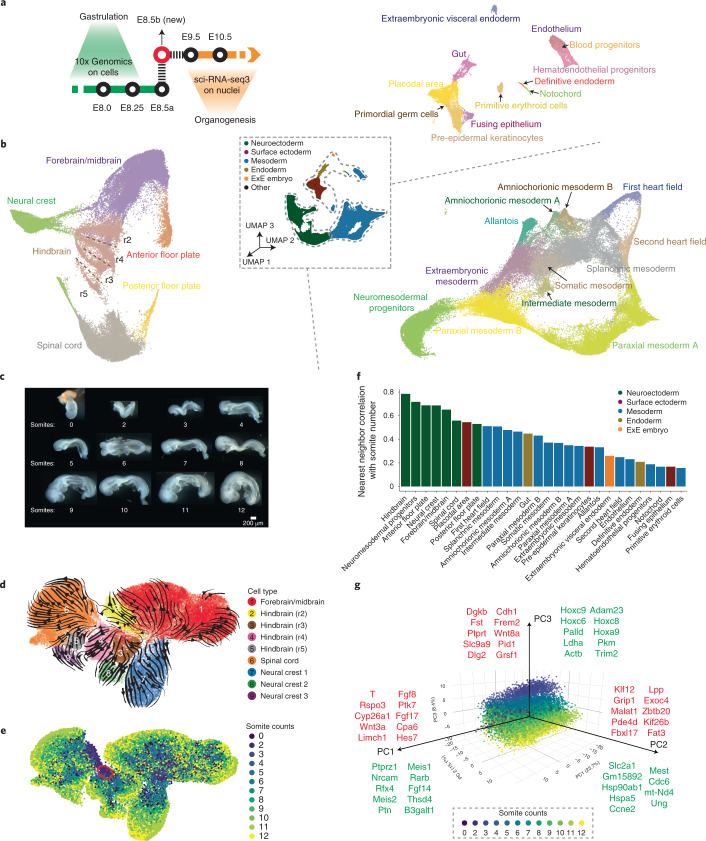
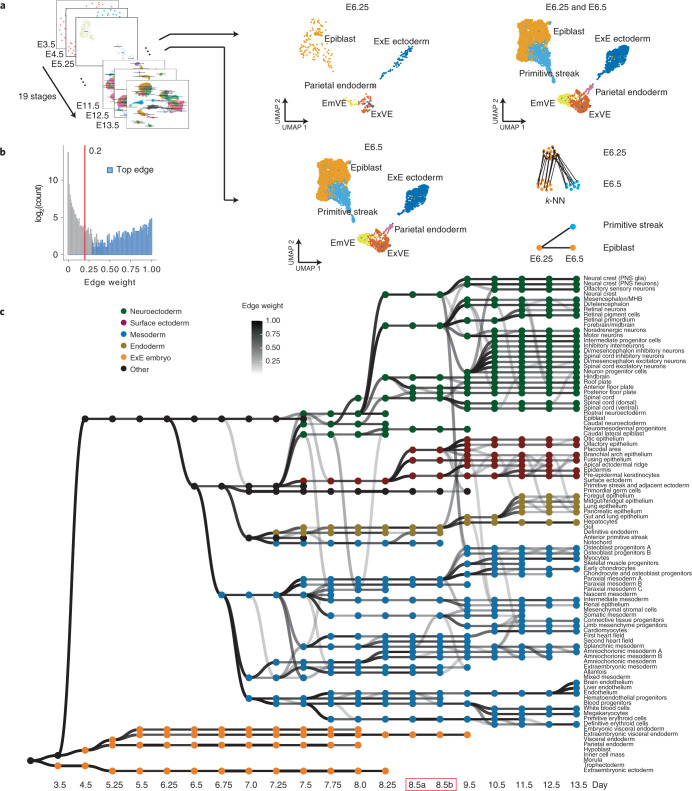
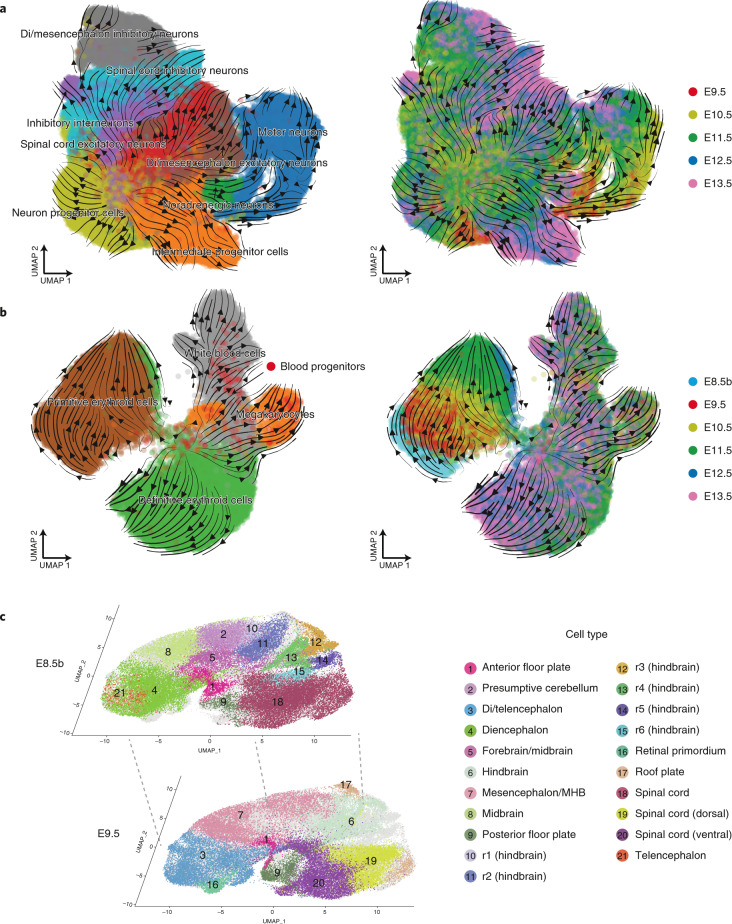
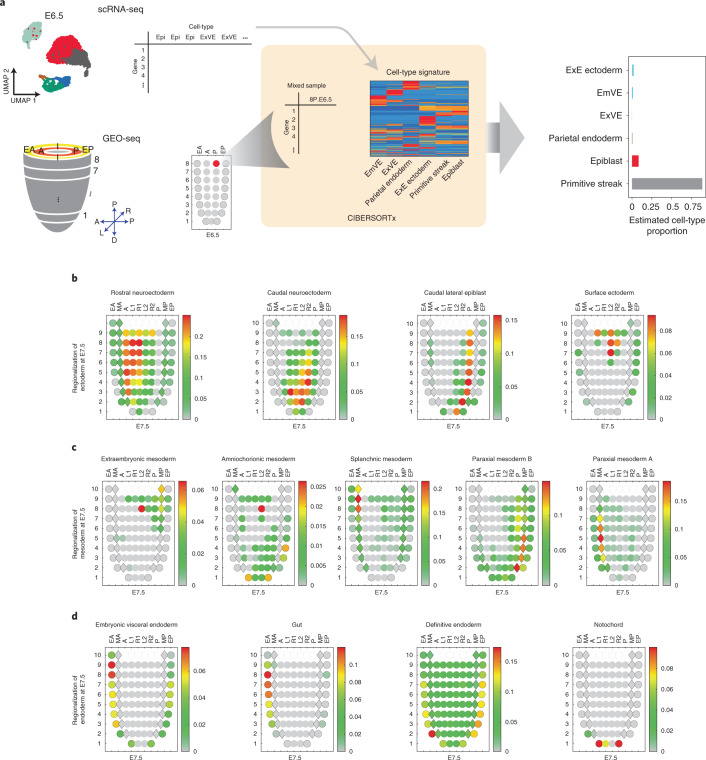
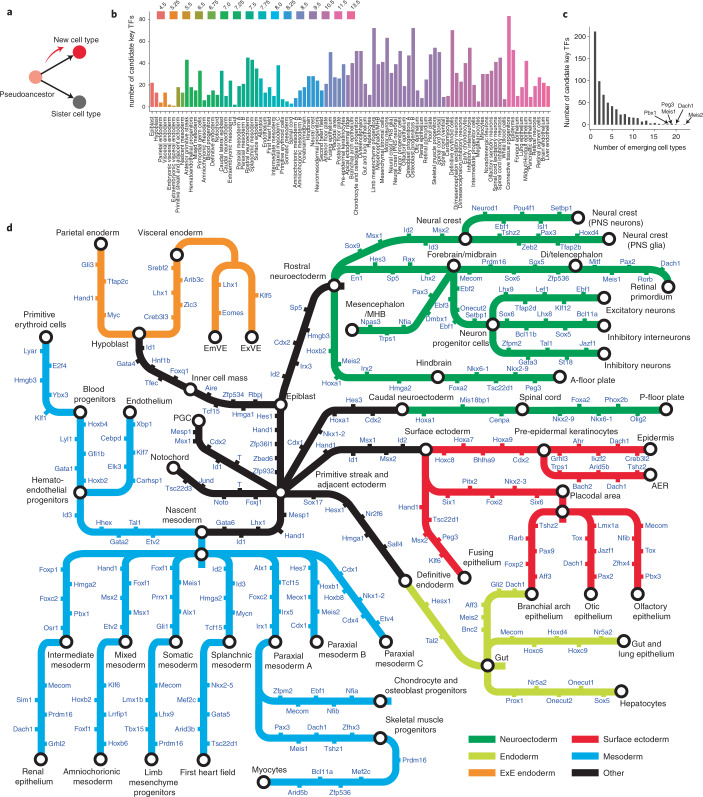
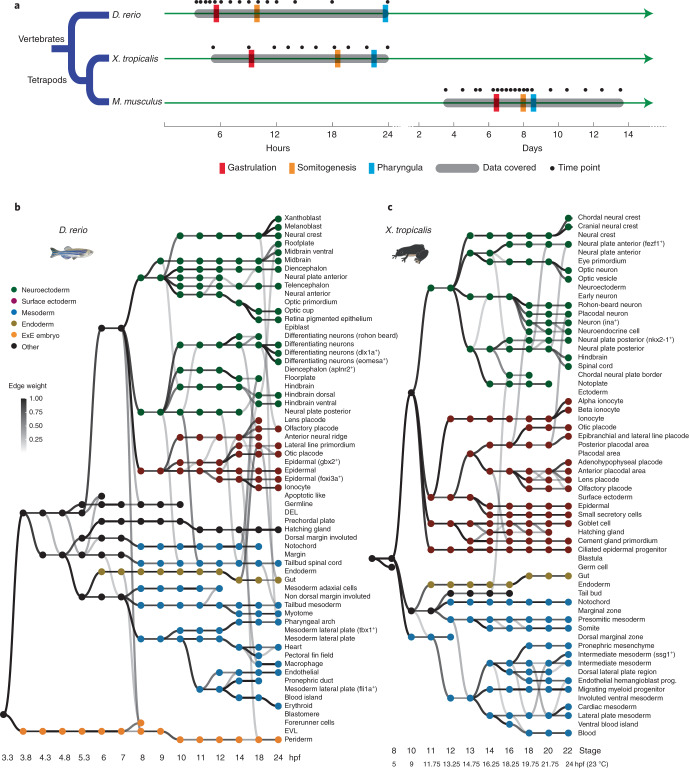
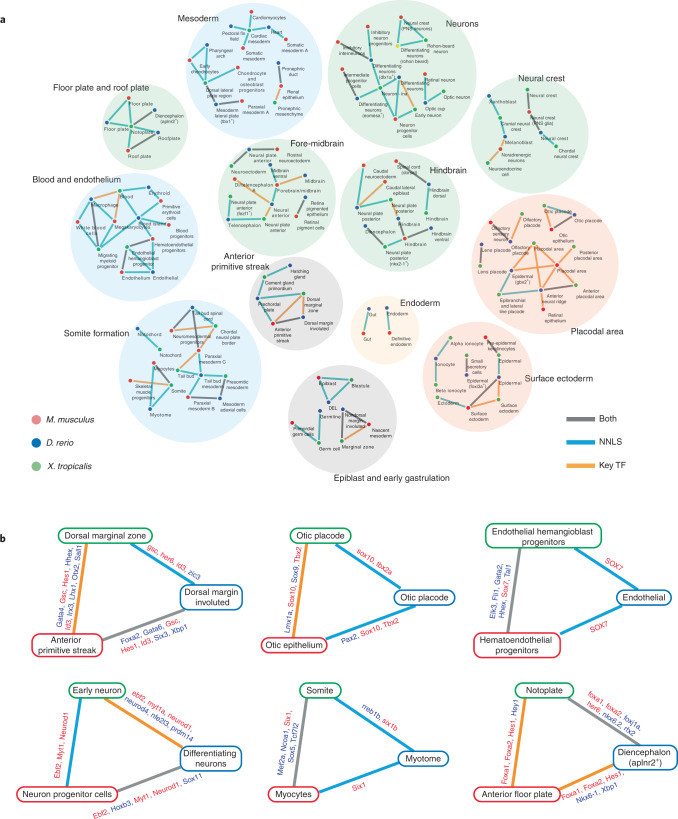
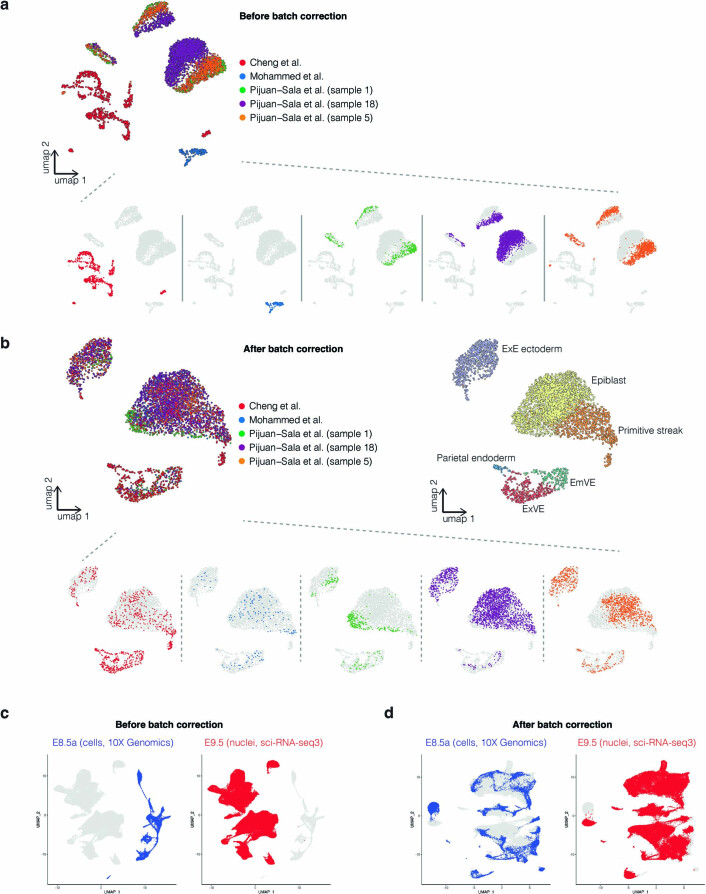
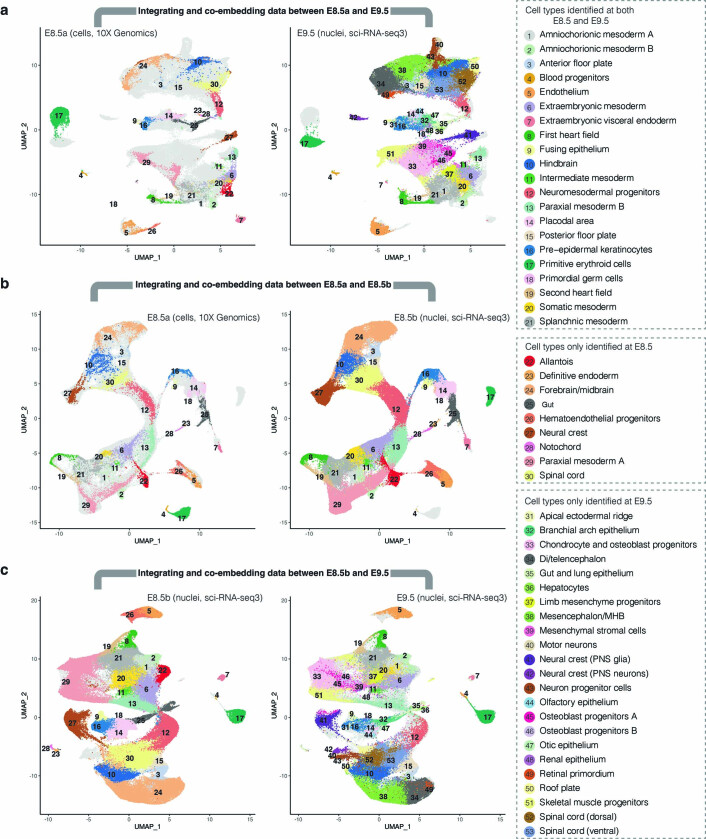
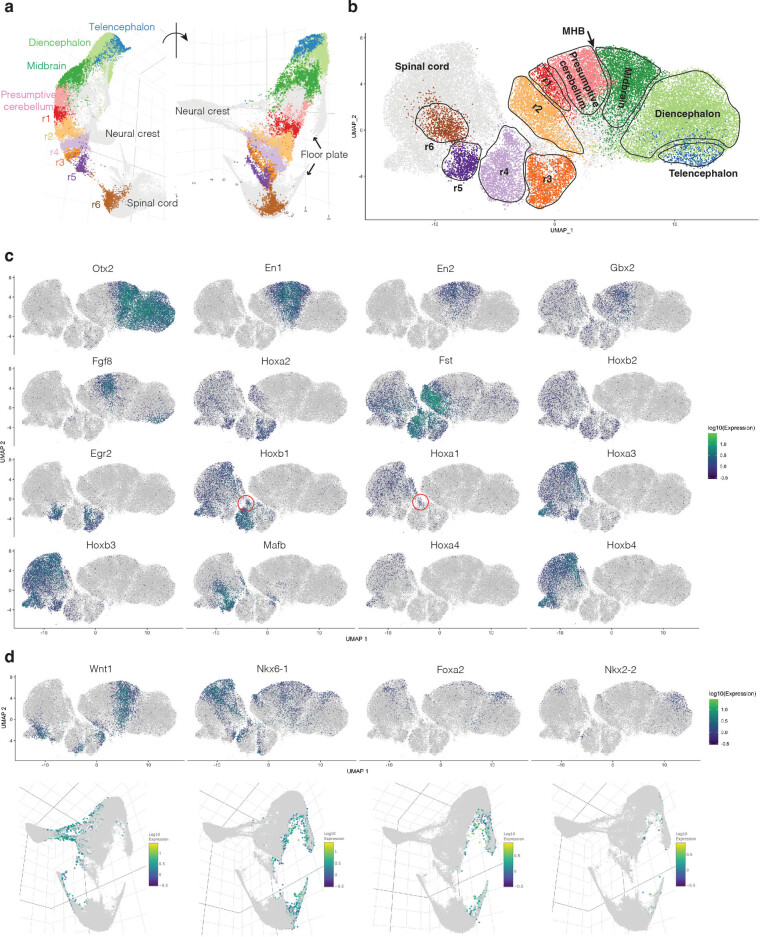
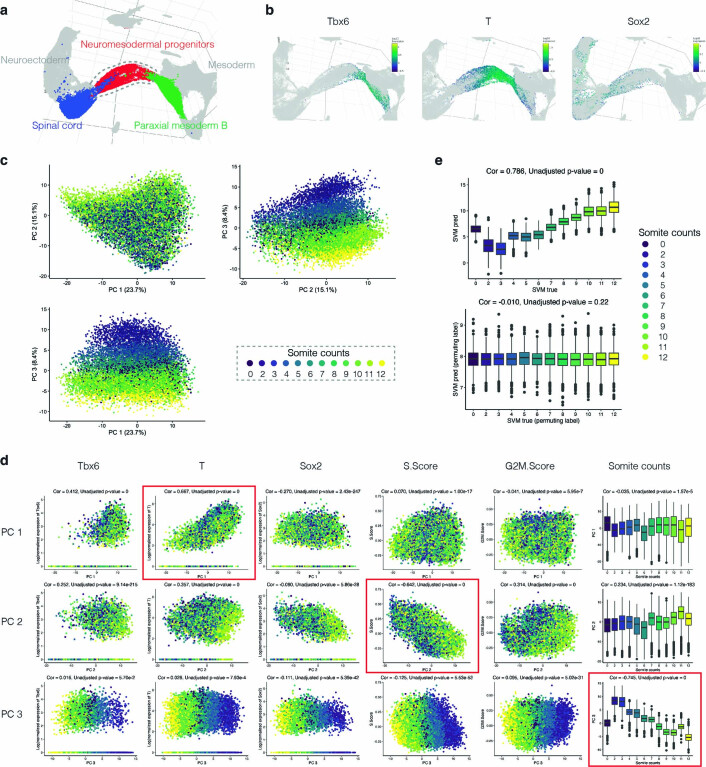
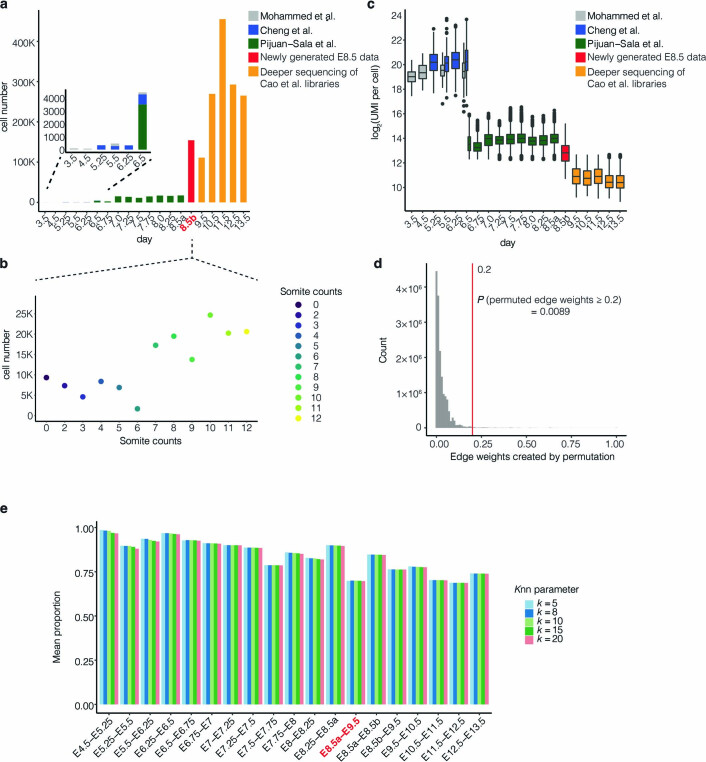
Mammalian embryogenesis is characterized by rapid cellular proliferation and diversification. Within a few weeks, a single-cell zygote gives rise to millions of cells expressing a panoply of molecular programs. Although intensively studied, a comprehensive delineation of the major cellular trajectories that comprise mammalian development in vivo remains elusive. Here, we set out to integrate several single-cell RNA-sequencing (scRNA-seq) datasets that collectively span mouse gastrulation and organogenesis, supplemented with new profiling of ~150,000 nuclei from approximately embryonic day 8.5 (E8.5) embryos staged in one-somite increments. Overall, we define cell states at each of 19 successive stages spanning E3.5 to E13.5 and heuristically connect them to their pseudoancestors and pseudodescendants. Although constructed through automated procedures, the resulting directed acyclic graph (TOME (trajectories of mammalian embryogenesis)) is largely consistent with our contemporary understanding of mammalian development. We leverage TOME to systematically nominate transcription factors (TFs) as candidate regulators of each cell type's specification, as well as 'cell-type homologs' across vertebrate evolution.
© 2022. The Author(s).
Conflict of interest statement
J.S. is a scientific advisory board member, consultant and/or cofounder of Cajal Neuroscience, Guardant Health, Maze Therapeutics, Camp4 Therapeutics, Phase Genomics, Adaptive Biotechnologies and Scale Biosciences. All other authors have no competing interests.
Figures

References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
