

Diagnostic accuracy of cerebrospinal fluid biomarkers in genetic prion diseases
- PMID: 35288744
- PMCID: PMC9014756
- DOI: 10.1093/brain/awab350
Diagnostic accuracy of cerebrospinal fluid biomarkers in genetic prion diseases
Abstract
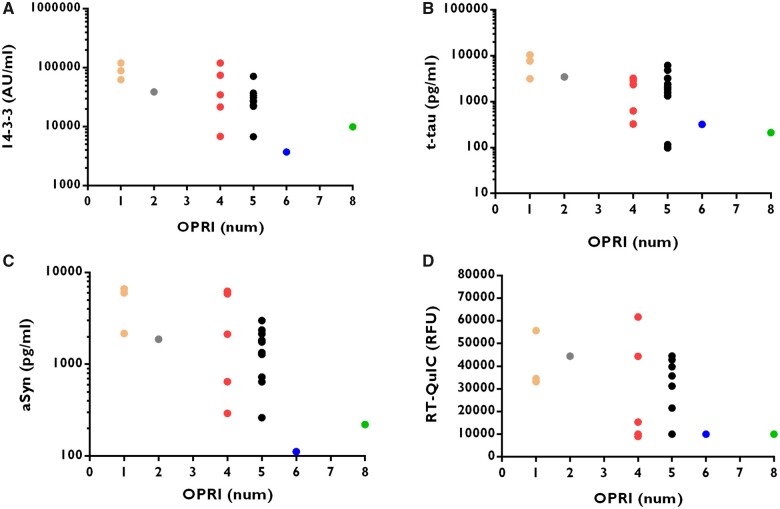
Genetic prion diseases are a rare and diverse group of fatal neurodegenerative disorders caused by pathogenic sequence variations in the prion protein gene, PRNP. Data on CSF biomarkers in patients with genetic prion diseases are limited and conflicting results have been reported for unclear reasons. Here, we aimed to analyse the diagnostic accuracy of CSF biomarkers currently used in prion clinical diagnosis in 302 symptomatic genetic prion disease cases from 11 prion diagnostic centres, encompassing a total of 36 different pathogenic sequence variations within the open reading frame of PRNP. CSF samples were assessed for the surrogate markers of neurodegeneration, 14-3-3 protein (14-3-3), total-tau protein (t-tau) and α-synuclein and for prion seeding activity through the real-time quaking-induced conversion assay. Biomarker results were compared with those obtained in healthy and neurological controls. For the most prevalent PRNP pathogenic sequence variations, biomarker accuracy and associations between biomarkers, demographic and genetic determinants were assessed. Additionally, the prognostic value of biomarkers for predicting total disease duration from symptom onset to death was investigated. High sensitivity of the four biomarkers was detected for genetic Creutzfeldt-Jakob disease associated with the E200K and V210I mutations, but low sensitivity was observed for mutations associated with Gerstmann-Sträussler-Scheinker syndrome and fatal familial insomnia. All biomarkers showed good to excellent specificity using the standard cut-offs often used for sporadic Creutzfeldt-Jakob disease. In genetic prion diseases related to octapeptide repeat insertions, the biomarker sensitivity correlated with the number of repeats. New genetic prion disease-specific cut-offs for 14-3-3, t-tau and α-synuclein were calculated. Disease duration in genetic Creutzfeldt-Jakob disease-E200K, Gerstmann-Sträussler-Scheinker-P102L and fatal familial insomnia was highly dependent on PRNP codon 129 MV polymorphism and was significantly associated with biomarker levels. In a large cohort of genetic prion diseases, the simultaneous analysis of CSF prion disease biomarkers allowed the determination of new mutation-specific cut-offs improving the discrimination of genetic prion disease cases and unveiled genetic prion disease-specific associations with disease duration.
Keywords: biomarker; cerebrospinal fluid; diagnostic marker; genetic prion diseases.
© The Author(s) (2022). Published by Oxford University Press on behalf of the Guarantors of Brain.
Figures


References
-
- Tee BL, Longoria Ibarrola EM, Geschwind MD. Prion Diseases. Neurol Clin. 2018;36(4):865–897. - PubMed
-
- Schmitz M, Dittmar K, Llorens F, et al. Hereditary human prion diseases: An update. Mol Neurobiol. 2017;54(6):4138–4112. - PubMed
-
- Gambetti P, Kong Q, Zou W, Parchi P, Chen SG.. Sporadic and familial CJD: Classification and characterisation. Br Med Bull. 2003;66:213–239. - PubMed
-
- Goldfarb LG, Petersen RB, Tabaton M, et al. Fatal familial insomnia and familial Creutzfeldt–Jakob disease: Disease phenotype determined by a DNA polymorphism. Science. 1992;258(5083):806–808. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
