

Spatial CRISPR genomics identifies regulators of the tumor microenvironment
- PMID: 35290801
- PMCID: PMC8992964
- DOI: 10.1016/j.cell.2022.02.015
Spatial CRISPR genomics identifies regulators of the tumor microenvironment
Abstract
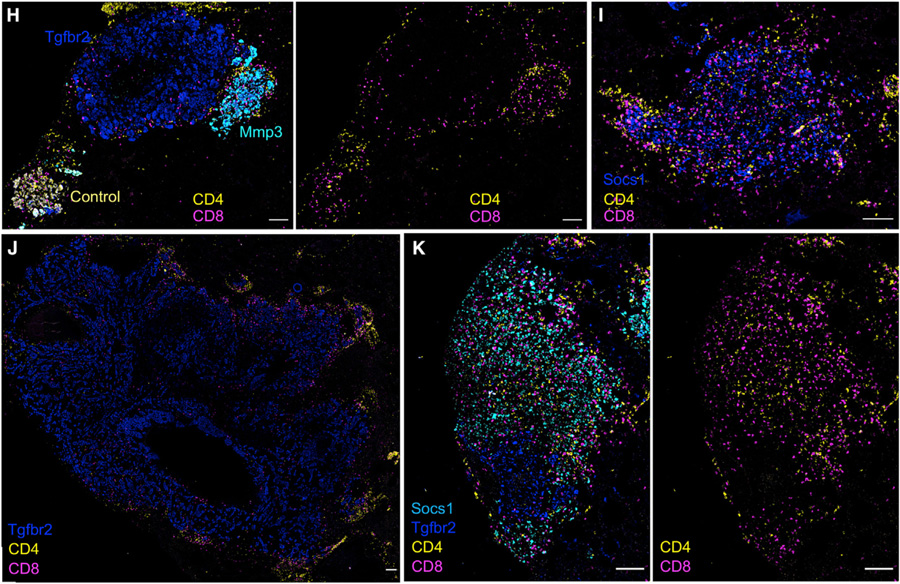
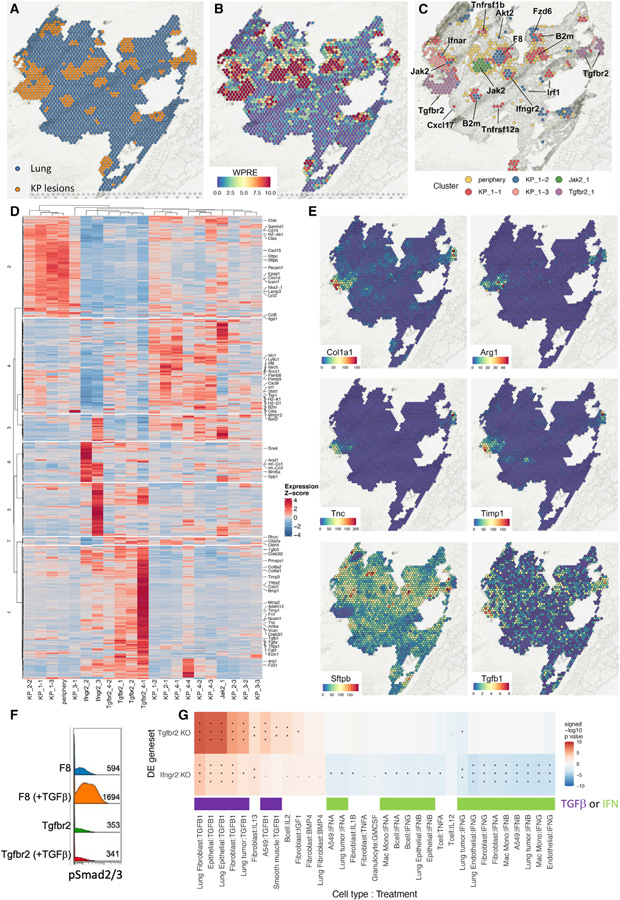
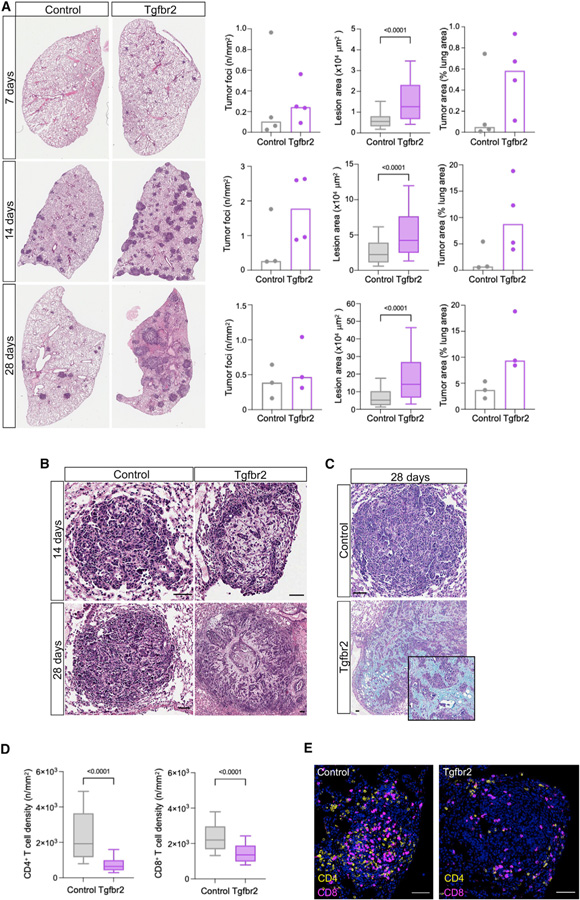
While CRISPR screens are helping uncover genes regulating many cell-intrinsic processes, existing approaches are suboptimal for identifying extracellular gene functions, particularly in the tissue context. Here, we developed an approach for spatial functional genomics called Perturb-map. We applied Perturb-map to knock out dozens of genes in parallel in a mouse model of lung cancer and simultaneously assessed how each knockout influenced tumor growth, histopathology, and immune composition. Moreover, we paired Perturb-map and spatial transcriptomics for unbiased analysis of CRISPR-edited tumors. We found that in Tgfbr2 knockout tumors, the tumor microenvironment (TME) was converted to a fibro-mucinous state, and T cells excluded, concomitant with upregulated TGFβ and TGFβ-mediated fibroblast activation, indicating that TGFβ-receptor loss on cancer cells increased TGFβ bioavailability and its immunosuppressive effects on the TME. These studies establish Perturb-map for functional genomics within the tissue at single-cell resolution with spatial architecture preserved and provide insight into how TGFβ responsiveness of cancer cells can affect the TME.
Keywords: CRISPR screens; Socs1; TGF beta; cancer immunology; interferon gamma; lung cancer; spatial genomics; spatial transcriptomics; tumor clonality; tumor microenvironment.
Copyright © 2022 Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests B.D.B. and A.W. have filed a patent application related to the Pro-Code technology.
Figures
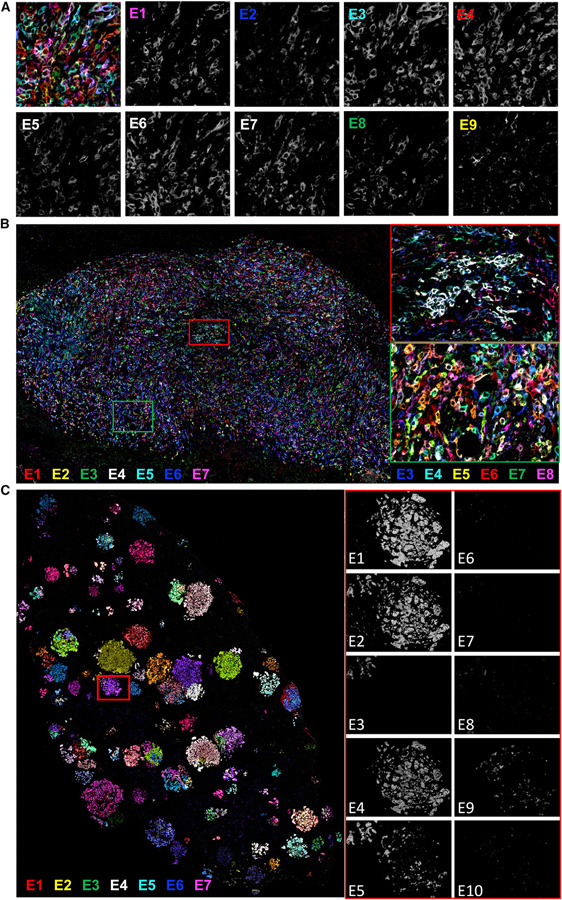
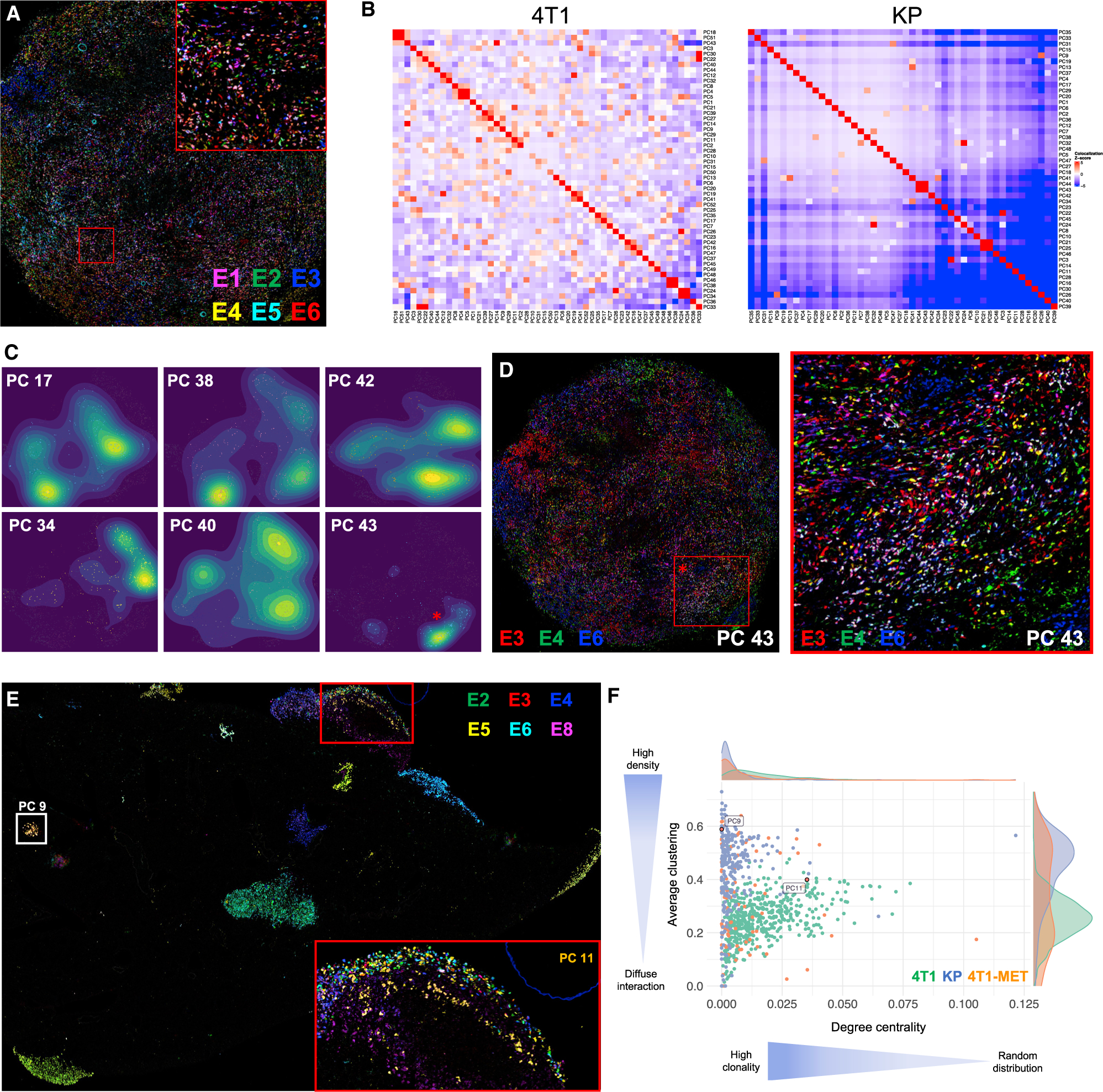
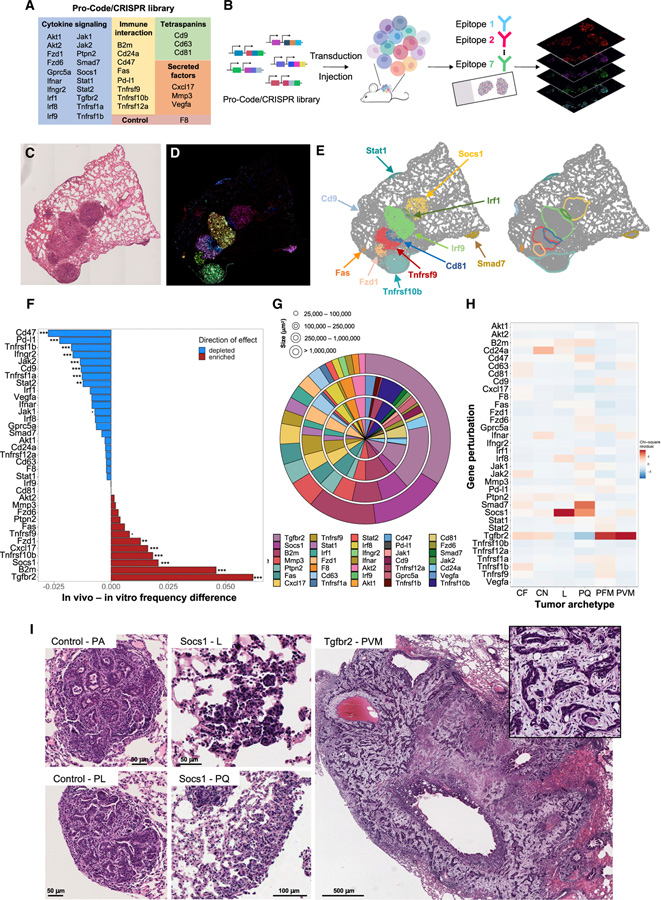
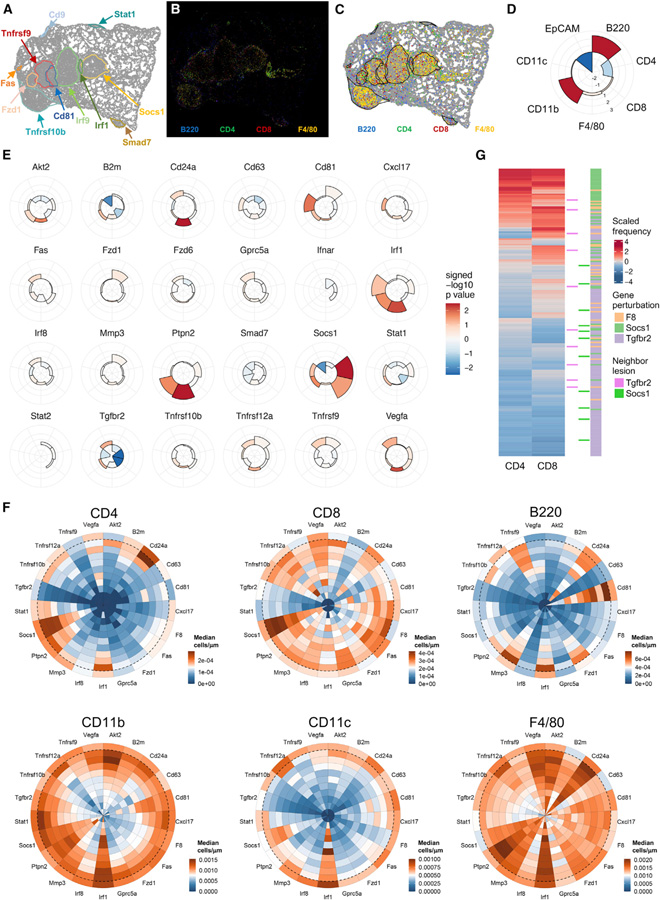
Comment in
-
Spatial CRISPR screens in tumors.Nat Methods. 2022 May;19(5):517. doi: 10.1038/s41592-022-01501-7. Nat Methods. 2022. PMID: 35545708 No abstract available.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
