

Immunopathology and Immunotherapy of Inflammatory Skin Diseases
- PMID: 35291649
- PMCID: PMC8901701
- DOI: 10.4110/in.2022.22.e7
Immunopathology and Immunotherapy of Inflammatory Skin Diseases
Abstract
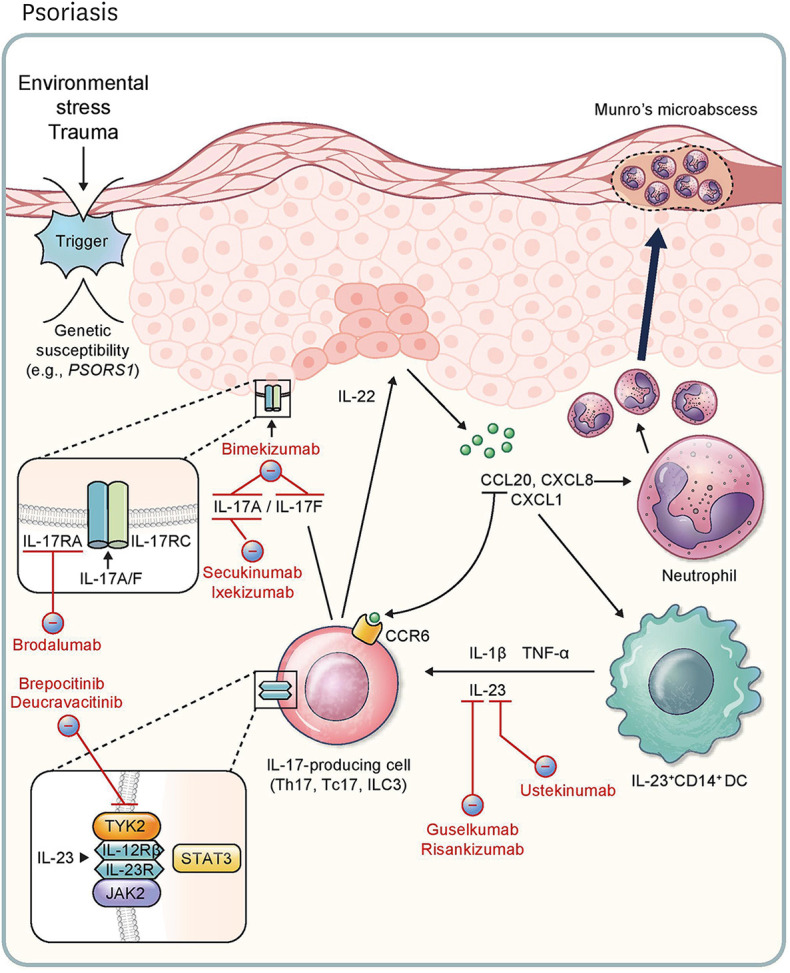
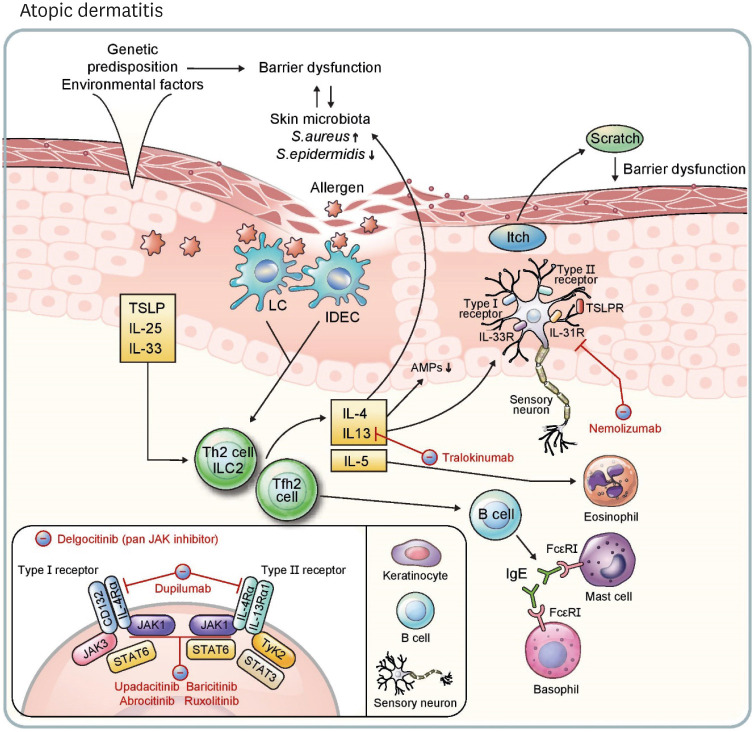
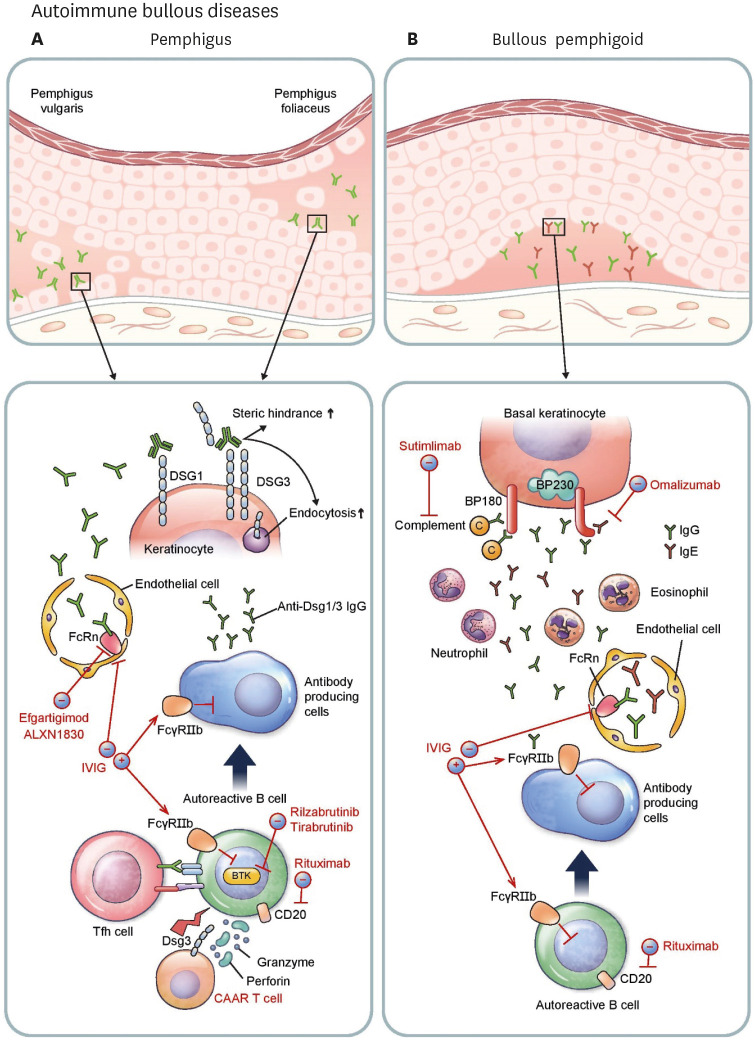
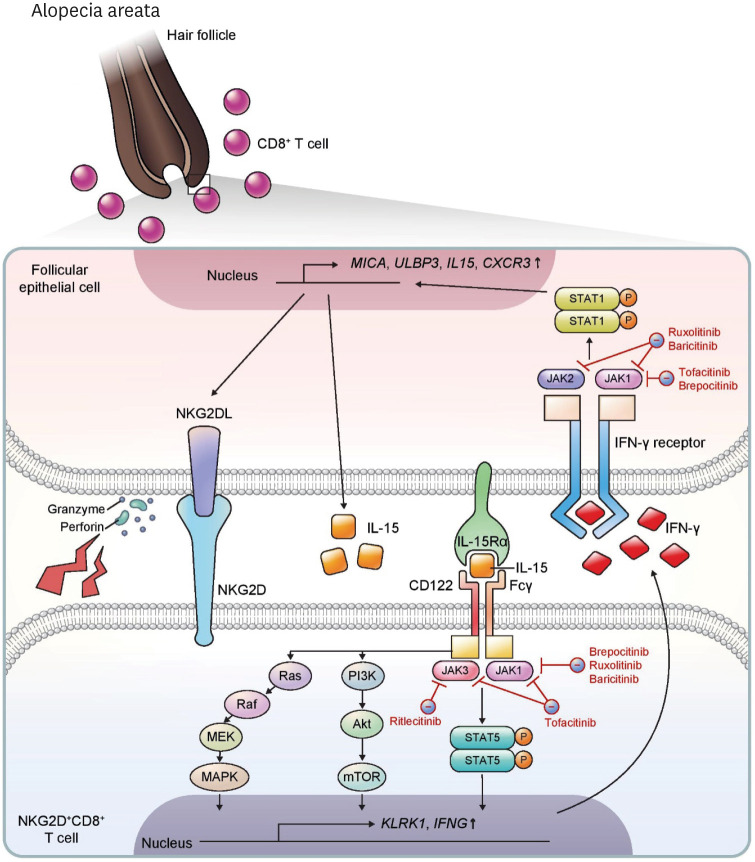
Recently, there have been impressive advancements in understanding of the immune mechanisms underlying cutaneous inflammatory diseases. To understand these diseases on a deeper level and clarify the therapeutic targets more precisely, numerous studies including in vitro experiments, animal models, and clinical trials have been conducted. This has resulted in a paradigm shift from non-specific suppression of the immune system to selective, targeted immunotherapies. These approaches target the molecular pathways and cytokines responsible for generating inflammatory conditions and reinforcing feedback mechanisms to aggravate inflammation. Among the numerous types of skin inflammation, psoriasis and atopic dermatitis (AD) are common chronic cutaneous inflammatory diseases. Psoriasis is a IL-17-mediated disease driven by IL-23, while AD is predominantly mediated by Th2 immunity. Autoimmune bullous diseases are autoantibody-mediated blistering disorders, including pemphigus and bullous pemphigoid. Alopecia areata is an organ-specific autoimmune disease mediated by CD8+ T-cells that targets hair follicles. This review will give an updated, comprehensive summary of the pathophysiology and immune mechanisms of inflammatory skin diseases. Moreover, the therapeutic potential of current and upcoming immunotherapies will be discussed.
Keywords: Alopecia areata; Atopic dermatitis; Biologics; Immunopathology; Pemphigoid, bullous; Pemphigus; Psoriasis.
Copyright © 2022. The Korean Association of Immunologists.
Conflict of interest statement
Conflicts of Interest: The authors declare no potential conflicts of interest.
Figures
References
-
- Greb JE, Goldminz AM, Elder JT, Lebwohl MG, Gladman DD, Wu JJ, Mehta NN, Finlay AY, Gottlieb AB. Psoriasis. Nat Rev Dis Primers. 2016;2:16082. - PubMed
-
- Alinaghi F, Calov M, Kristensen LE, Gladman DD, Coates LC, Jullien D, Gottlieb AB, Gisondi P, Wu JJ, Thyssen JP, et al. Prevalence of psoriatic arthritis in patients with psoriasis: a systematic review and meta-analysis of observational and clinical studies. J Am Acad Dermatol. 2019;80:251–265.e219. - PubMed
-
- Heydendael VM, Spuls PI, Opmeer BC, de Borgie CA, Reitsma JB, Goldschmidt WF, Bossuyt PM, Bos JD, de Rie MA. Methotrexate versus cyclosporine in moderate-to-severe chronic plaque psoriasis. N Engl J Med. 2003;349:658–665. - PubMed
Publication types
LinkOut - more resources
Full Text Sources
Research Materials
