

Origin of thiocyanate spectral shifts in water and organic solvents
- PMID: 35291777
- PMCID: PMC8923707
- DOI: 10.1063/5.0082969
Origin of thiocyanate spectral shifts in water and organic solvents
Abstract
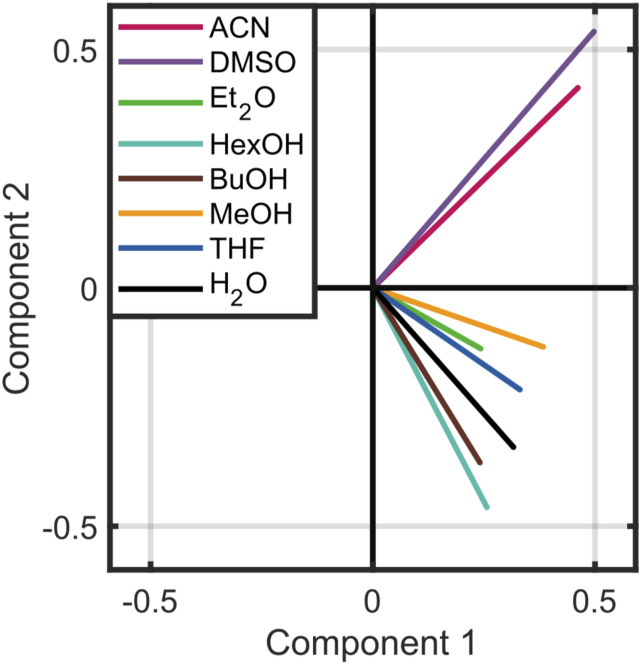
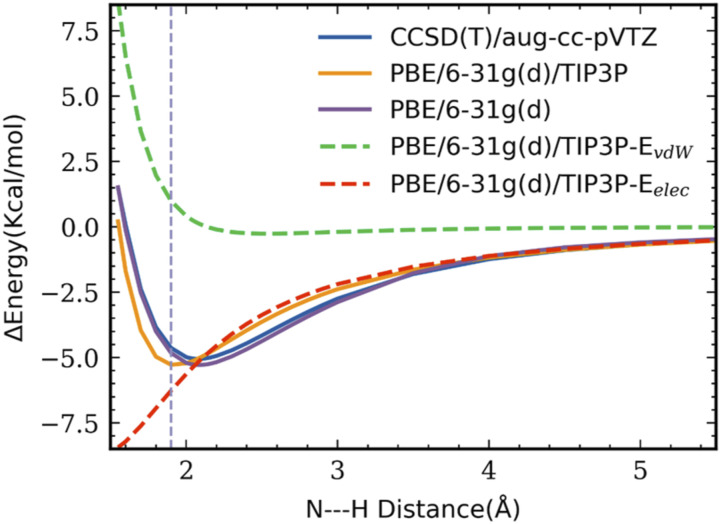
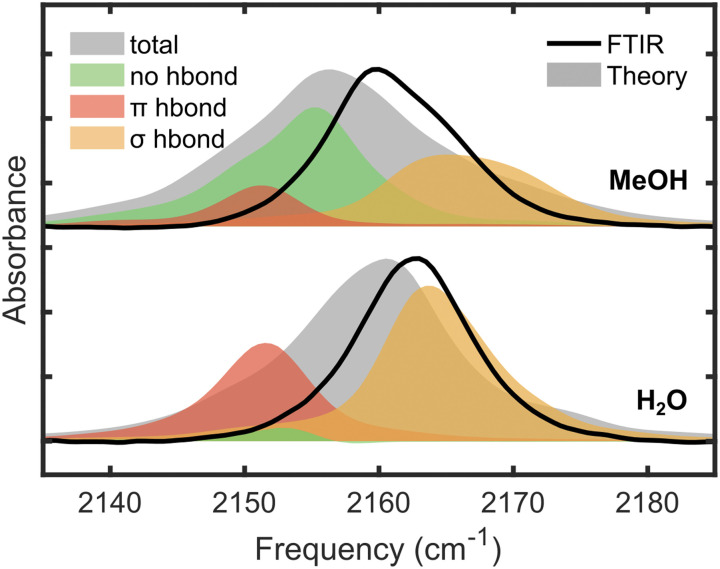
Vibrational spectroscopy is a useful technique for probing chemical environments. The development of models that can reproduce the spectra of nitriles and azides is valuable because these probes are uniquely suited for investigating complex systems. Empirical vibrational spectroscopic maps are commonly employed to obtain the instantaneous vibrational frequencies during molecular dynamics simulations but often fail to adequately describe the behavior of these probes, especially in its transferability to a diverse range of environments. In this paper, we demonstrate several reasons for the difficulty in constructing a general-purpose vibrational map for methyl thiocyanate (MeSCN), a model for cyanylated biological probes. In particular, we found that electrostatics alone are not a sufficient metric to categorize the environments of different solvents, and the dominant features in intermolecular interactions in the energy landscape vary from solvent to solvent. Consequently, common vibrational mapping schemes do not cover all essential interaction terms adequately, especially in the treatment of van der Waals interactions. Quantum vibrational perturbation (QVP) theory, along with a combined quantum mechanical and molecular mechanical potential for solute-solvent interactions, is an alternative and efficient modeling technique, which is compared in this paper, to yield spectroscopic results in good agreement with experimental FTIR. QVP has been used to analyze the computational data, revealing the shortcomings of the vibrational maps for MeSCN in different solvents. The results indicate that insights from QVP analysis can be used to enhance the transferability of vibrational maps in future studies.
Figures






References
-
- Chabal Y. J., Surf. Sci. Rep. 8, 211 (1988).10.1016/0167-5729(88)90011-8 - DOI
-
- L. G. Wade, Jr., Organic Chemistry, 8th ed. (Pearson Education, Inc., 2016).
Grants and funding
LinkOut - more resources
Full Text Sources