

Swapping Metagenomics Preprocessing Pipeline Components Offers Speed and Sensitivity Increases
- PMID: 35293792
- PMCID: PMC9040843
- DOI: 10.1128/msystems.01378-21
Swapping Metagenomics Preprocessing Pipeline Components Offers Speed and Sensitivity Increases
Abstract
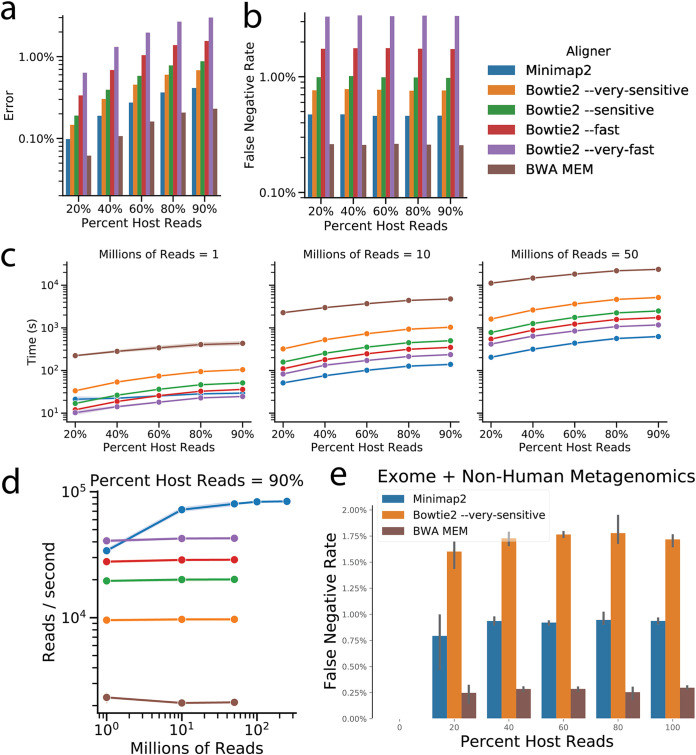
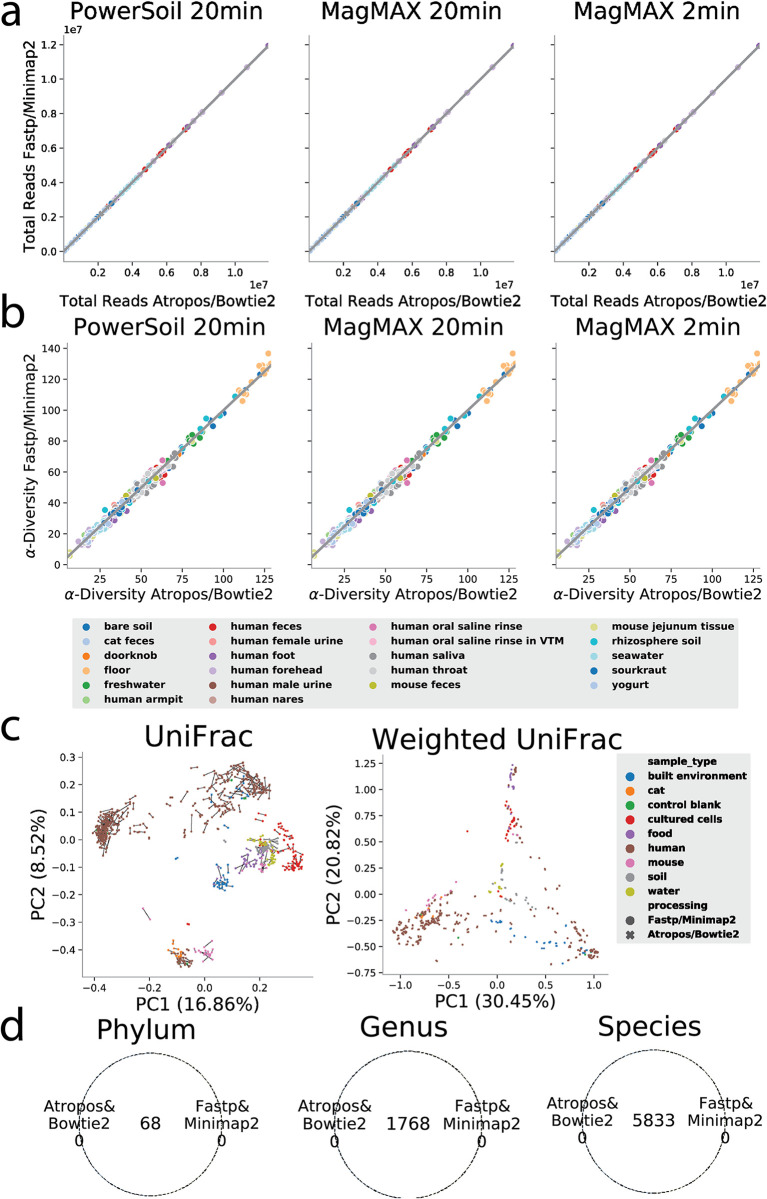
Increasing data volumes on high-throughput sequencing instruments such as the NovaSeq 6000 leads to long computational bottlenecks for common metagenomics data preprocessing tasks such as adaptor and primer trimming and host removal. Here, we test whether faster recently developed computational tools (Fastp and Minimap2) can replace widely used choices (Atropos and Bowtie2), obtaining dramatic accelerations with additional sensitivity and minimal loss of specificity for these tasks. Furthermore, the taxonomic tables resulting from downstream processing provide biologically comparable results. However, we demonstrate that for taxonomic assignment, Bowtie2's specificity is still required. We suggest that periodic reevaluation of pipeline components, together with improvements to standardized APIs to chain them together, will greatly enhance the efficiency of common bioinformatics tasks while also facilitating incorporation of further optimized steps running on GPUs, FPGAs, or other architectures. We also note that a detailed exploration of available algorithms and pipeline components is an important step that should be taken before optimization of less efficient algorithms on advanced or nonstandard hardware. IMPORTANCE In shotgun metagenomics studies that seek to relate changes in microbial DNA across samples, processing the data on a computer often takes longer than obtaining the data from the sequencing instrument. Recently developed software packages that perform individual steps in the pipeline of data processing in principle offer speed advantages, but in practice they may contain pitfalls that prevent their use, for example, they may make approximations that introduce unacceptable errors in the data. Here, we show that differences in choices of these components can speed up overall data processing by 5-fold or more on the same hardware while maintaining a high degree of correctness, greatly reducing the time taken to interpret results. This is an important step for using the data in clinical settings, where the time taken to obtain the results may be critical for guiding treatment.
Keywords: alignment; host filtering; metagenomics.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Shaffer JP, Marotz C, Belda-Ferre P, Martino C, Wandro S, Estaki M, Salido RA, Carpenter CS, Zaramela LS, Minich JJ, Bryant M, Sanders K, Fraraccio S, Ackermann G, Humphrey G, Swafford AD, Miller-Montgomery S, Knight R. 2021. A comparison of DNA/RNA extraction protocols for high-throughput sequencing of microbial communities. Biotechniques 70:149–159. doi:10.2144/btn-2020-0153. - DOI - PMC - PubMed
-
- Salosensaari A, Laitinen V, Havulinna AS, Meric G, Cheng S, Perola M, Valsta L, Alfthan G, Inouye M, Watrous JD, Long T, Salido RA, Sanders K, Brennan C, Humphrey GC, Sanders JG, Jain M, Jousilahti P, Salomaa V, Knight R, Lahti L, Niiranen T. 2021. Taxonomic signatures of cause-specific mortality risk in human gut microbiome. Nat Commun 12:2671. doi:10.1038/s41467-021-22962-y. - DOI - PMC - PubMed
