

RosettaSurf-A surface-centric computational design approach
- PMID: 35294435
- PMCID: PMC9015148
- DOI: 10.1371/journal.pcbi.1009178
RosettaSurf-A surface-centric computational design approach
Abstract
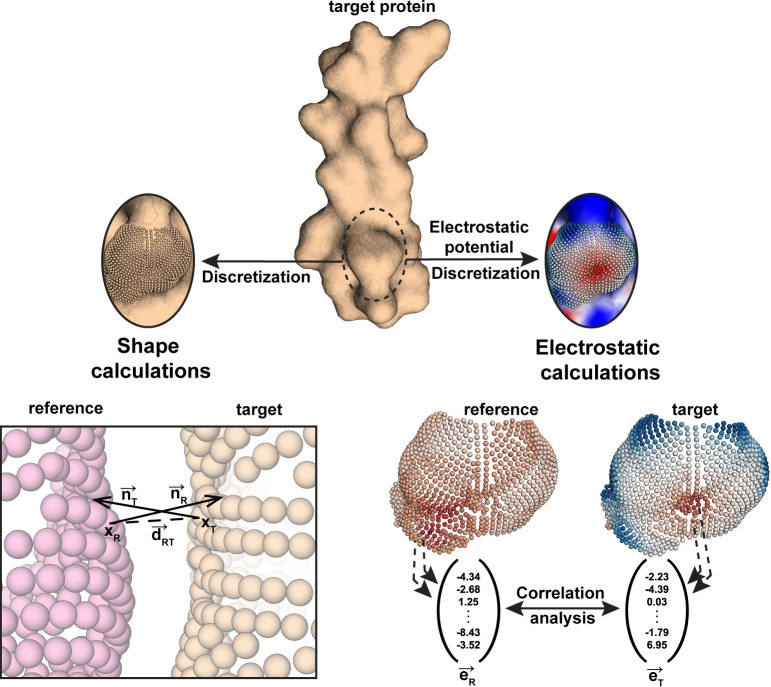
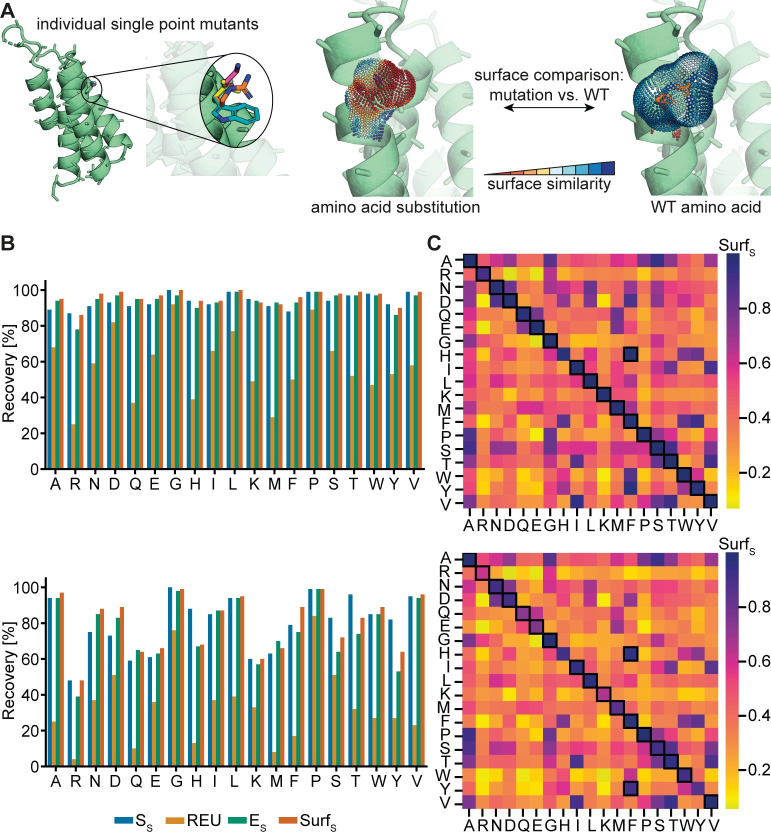
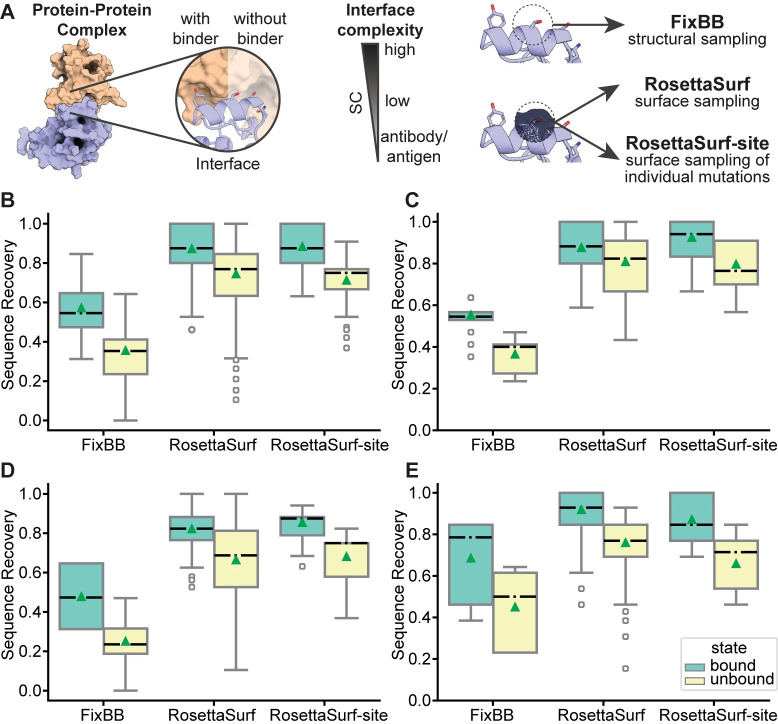
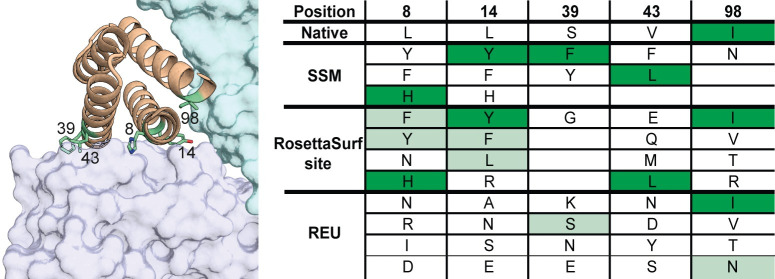
Proteins are typically represented by discrete atomic coordinates providing an accessible framework to describe different conformations. However, in some fields proteins are more accurately represented as near-continuous surfaces, as these are imprinted with geometric (shape) and chemical (electrostatics) features of the underlying protein structure. Protein surfaces are dependent on their chemical composition and, ultimately determine protein function, acting as the interface that engages in interactions with other molecules. In the past, such representations were utilized to compare protein structures on global and local scales and have shed light on functional properties of proteins. Here we describe RosettaSurf, a surface-centric computational design protocol, that focuses on the molecular surface shape and electrostatic properties as means for protein engineering, offering a unique approach for the design of proteins and their functions. The RosettaSurf protocol combines the explicit optimization of molecular surface features with a global scoring function during the sequence design process, diverging from the typical design approaches that rely solely on an energy scoring function. With this computational approach, we attempt to address a fundamental problem in protein design related to the design of functional sites in proteins, even when structurally similar templates are absent in the characterized structural repertoire. Surface-centric design exploits the premise that molecular surfaces are, to a certain extent, independent of the underlying sequence and backbone configuration, meaning that different sequences in different proteins may present similar surfaces. We benchmarked RosettaSurf on various sequence recovery datasets and showcased its design capabilities by generating epitope mimics that were biochemically validated. Overall, our results indicate that the explicit optimization of surface features may lead to new routes for the design of functional proteins.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
References
-
- Connolly ML. Analytical molecular surface calculation. Journal of applied crystallography. 1983;16(5):548–58.
-
- Richards FM. Areas, Volumes, Packing, and Protein Structure. Annu Rev Biophys Bioeng. 1977. Jun;6(1):151–76. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
