

Loss of mouse Stmn2 function causes motor neuropathy
- PMID: 35294901
- PMCID: PMC9119928
- DOI: 10.1016/j.neuron.2022.02.011
Loss of mouse Stmn2 function causes motor neuropathy
Erratum in
-
Loss of mouse Stmn2 function causes motor neuropathy.Neuron. 2022 Dec 7;110(23):4031. doi: 10.1016/j.neuron.2022.11.003. Neuron. 2022. PMID: 36480942 No abstract available.
Abstract
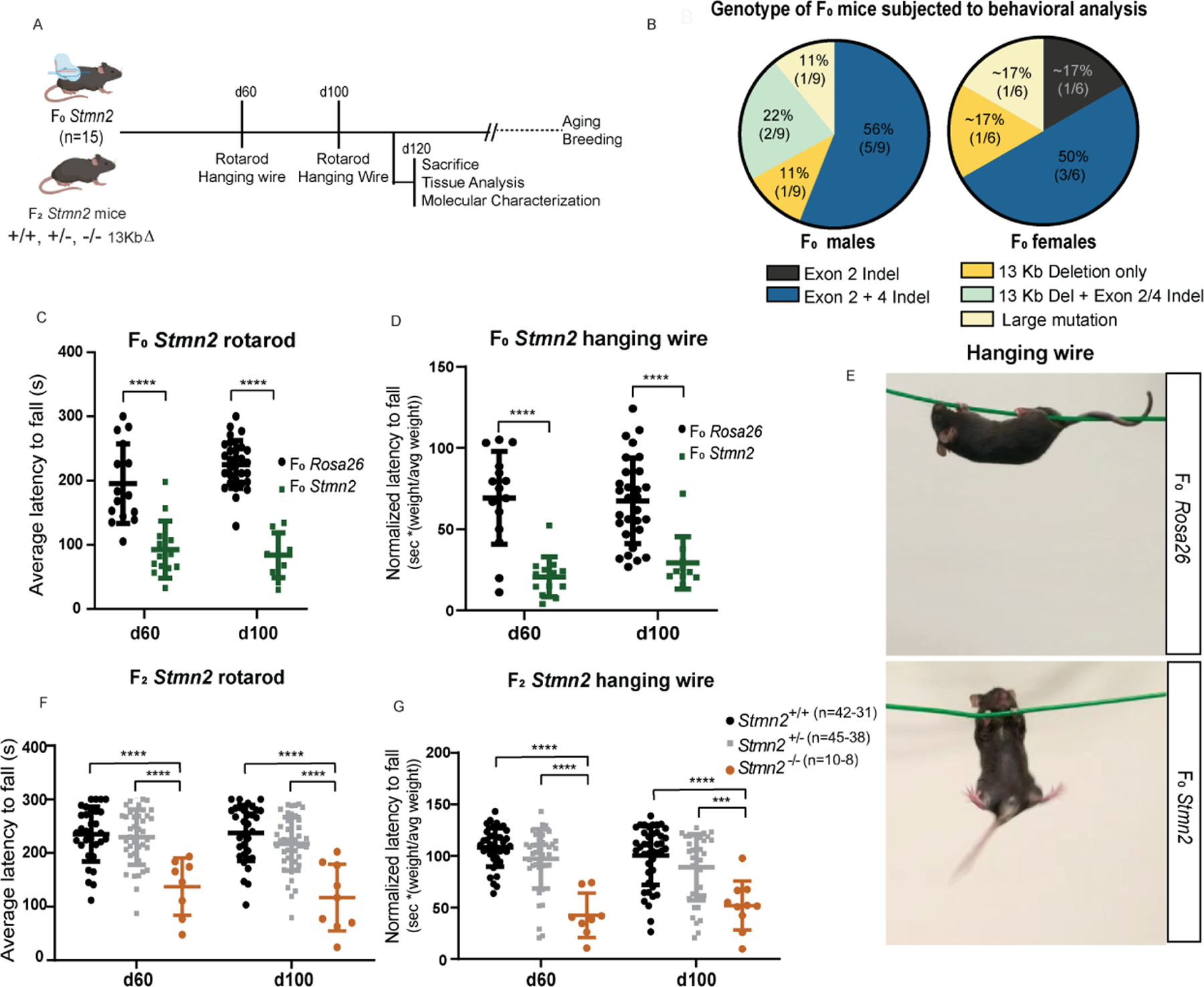
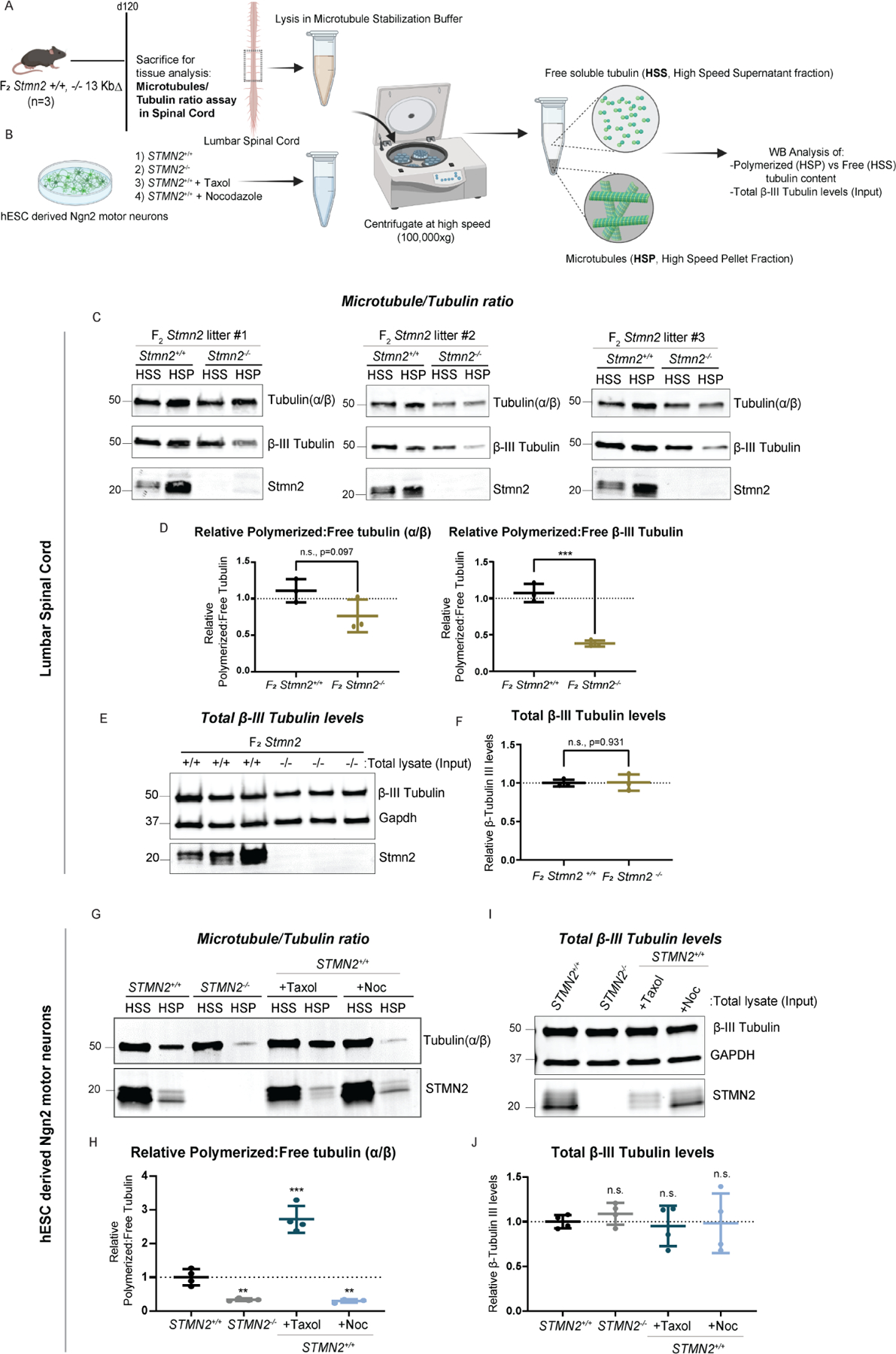
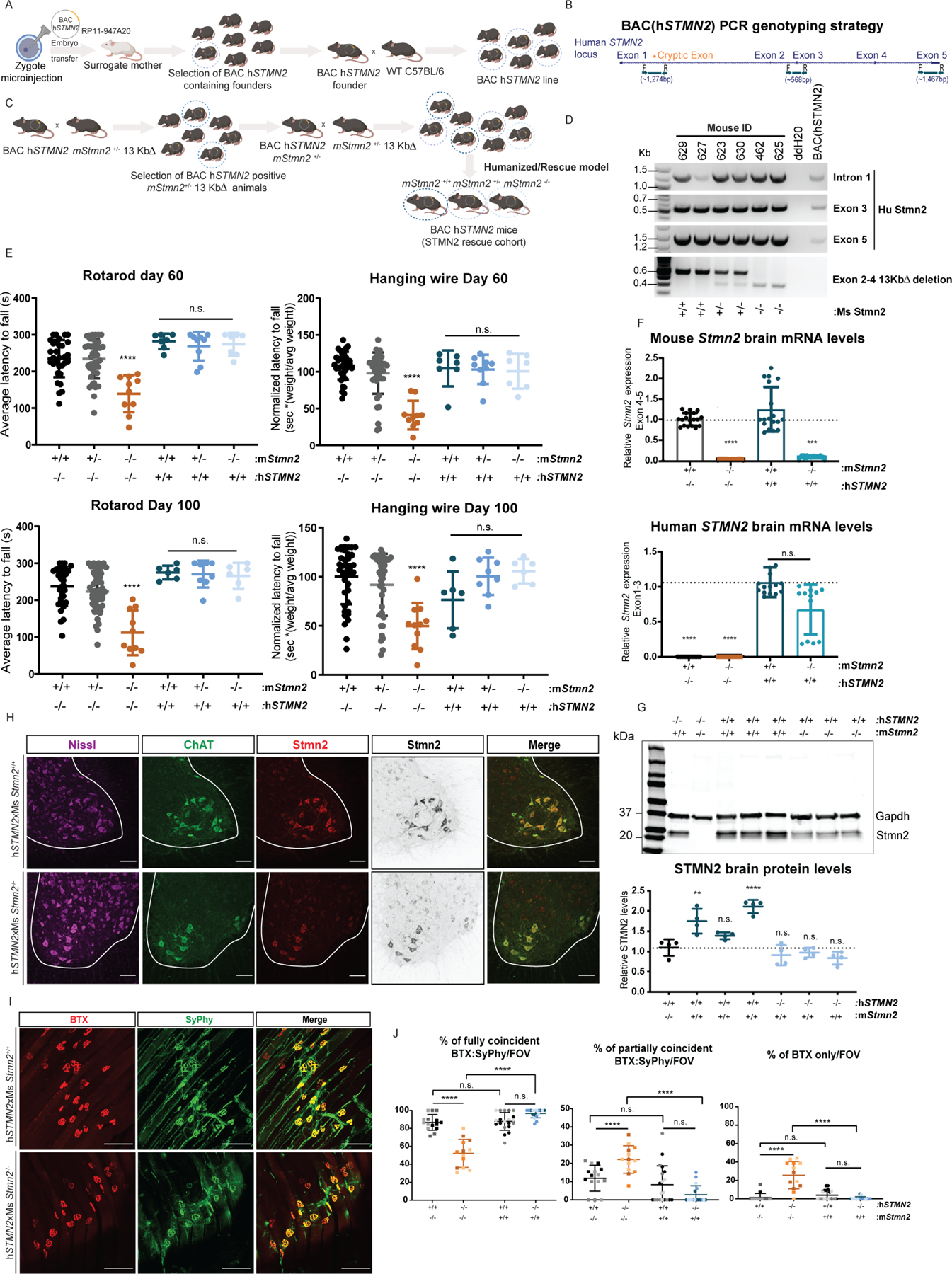
Amyotrophic lateral sclerosis (ALS) is characterized by motor neuron degeneration accompanied by aberrant accumulation and loss of function of the RNA-binding protein TDP43. Thus far, it remains unresolved to what extent TDP43 loss of function directly contributes to motor system dysfunction. Here, we employed gene editing to find whether the mouse ortholog of the TDP43-regulated gene STMN2 has an important function in maintaining the motor system. Both mosaic founders and homozygous loss-of-function Stmn2 mice exhibited neuromuscular junction denervation and fragmentation, resulting in muscle atrophy and impaired motor behavior, accompanied by an imbalance in neuronal microtubule dynamics in the spinal cord. The introduction of human STMN2 through BAC transgenesis was sufficient to rescue the motor phenotypes observed in Stmn2 mutant mice. Collectively, our results demonstrate that disrupting the ortholog of a single TDP43-regulated RNA is sufficient to cause substantial motor dysfunction, indicating that disruption of TDP43 function is likely a contributor to ALS.
Keywords: ALS; CRISPR; SCG10; TARDBP; TDP43; microtubules; motor neuron; motor neuropathy; neuromuscular junction; stathmin 2.
Copyright © 2022 Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests K.E. is a cofounder of Q-State Biosciences, Quralis, and Enclear Therapies and currently head of research and early development at BioMarin Pharmaceutical. J.R.K. is an employee of Faze Medicines and a shareholder in Faze Medicines and QurAlis. K.E., I.G.S.J., J.R.K., and F.L. are authors on a pending patent that describes methods and compositions for restoring STMN2 levels (WO/2020/150290). K.E. is an author on a pending patent that describes compounds and methods for treating neurodegenerative diseases (WO2020107037).
Figures





References
-
- Al-Chalabi A, Calvo A, Chio A, Colville S, Ellis CM, Hardiman O, Heverin M, Howard RS, Huisman MHB, Keren N, et al. (2014). Analysis of amyotrophic lateral sclerosis as a multistep process: a population-based modelling study. Lancet Neurol 13, 1108–1113. 10.1016/S14744422(14)70219-4. - DOI - PMC - PubMed
-
- Arnold ES, Ling SC, Huelga SC, Lagier-Tourenne C, Polymenidou M, Ditsworth D, Kordasiewicz HB, McAlonis-Downes M, Platoshyn O, Parone PA, et al. (2013). ALS-linked TDP-43 mutations produce aberrant RNA splicing and adult-onset motor neuron disease without aggregation or loss of nuclear TDP-43. Proceedings of the National Academy of Sciences of the United States of America 110, E736–745. 10.1073/pnas.1222809110 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Miscellaneous
