

Mortality and survival in idiopathic pulmonary fibrosis: a systematic review and meta-analysis
- PMID: 35295232
- PMCID: PMC8918939
- DOI: 10.1183/23120541.00591-2021
Mortality and survival in idiopathic pulmonary fibrosis: a systematic review and meta-analysis
Abstract
Background: There are substantial advances in diagnosis and treatment for idiopathic pulmonary fibrosis (IPF), but without much evidence available on recent mortality and survival trends.
Methods: A narrative synthesis approach was used to investigate the mortality trends, then meta-analyses for survival trends were carried out based on various time periods.
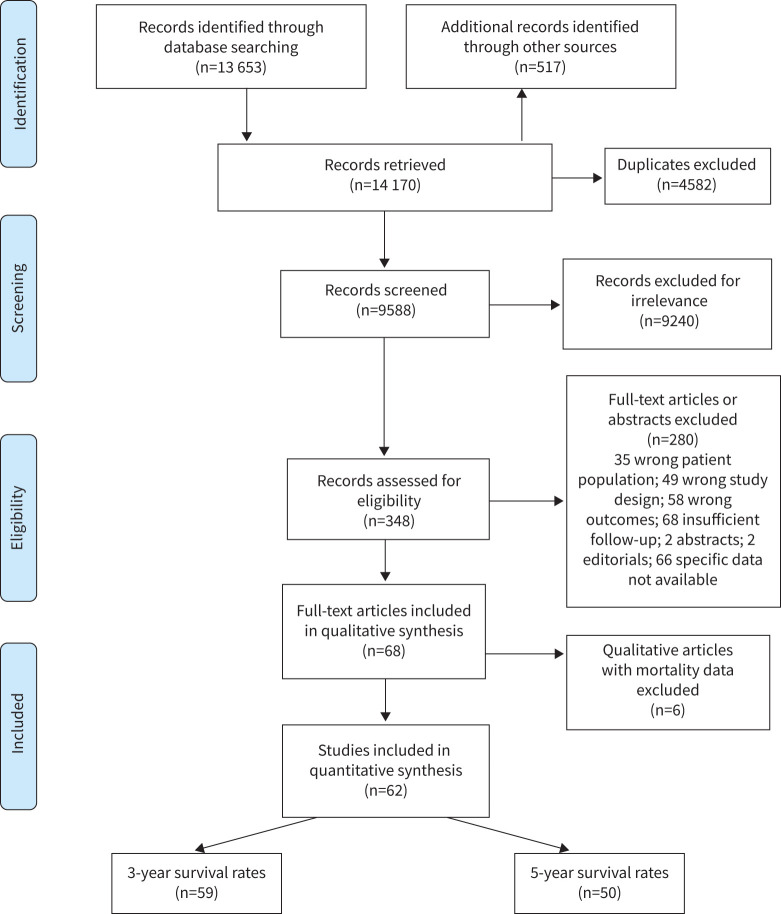
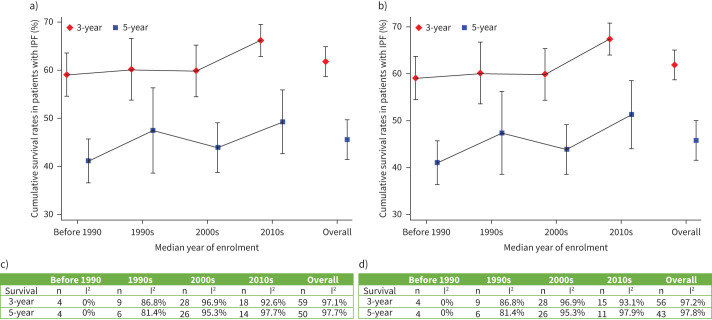
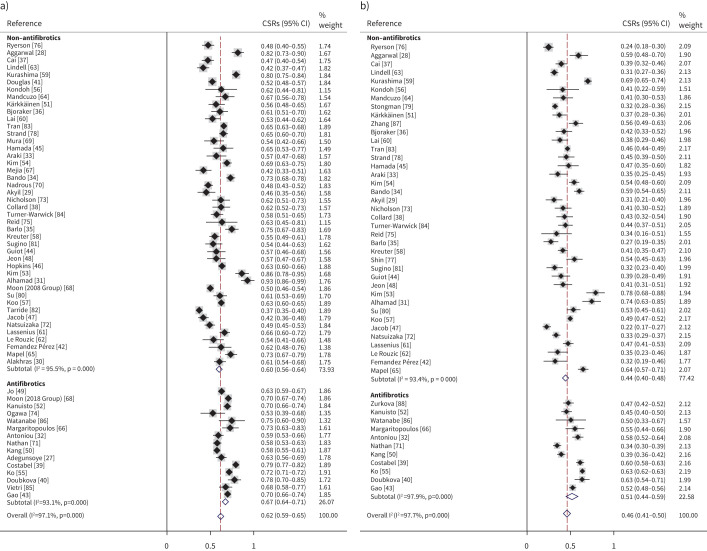
Results: Six studies reported the mortality data for IPF in 22 countries, and 62 studies (covering 63 307 patients from 20 countries) reported survival data for IPF. Age-standardised mortality for IPF varied from ∼0.5 to ∼12 per 100 000 population per year after year 2000. There were increased mortality trends for IPF in Australia, Brazil, Belgium, Canada, Czech Republic, Finland, France, Germany, Hungary, Italy, Lithuania, the Netherlands, Poland, Portugal, Spain, Sweden and UK, while Austria, Croatia, Denmark, Romania and the USA showed decreased mortality trends. The overall 3-year and 5-year cumulative survival rates (CSRs) were 61.8% (95% CI 58.7-64.9; I2=97.1%) and 45.6% (95% CI 41.5-49.7; I2=97.7%), respectively. Prior to 2010, the pooled 3-year CSR was 59.9% (95% CI 55.8-64.1; I2=95.8%), then not significantly (p=0.067) increased to 66.2% (95% CI 62.9-69.5; I2=92.6%) in the 2010s decade. After excluding three studies in which no patients received antifibrotics after year 2010, the pooled 3-year CSRs significantly (p=0.039) increased to 67.4% (95% CI 63.9-70.9; I2=93.1%) in the 2010s decade.
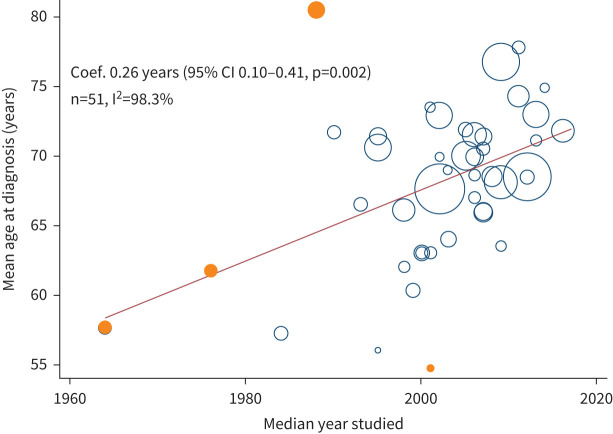
Discussion: IPF is a diagnosis associated with high mortality. There was no observed increasing survival trend for patients with IPF before year 2010, with then a switch to an improvement, which is probably multifactorial.
Copyright ©The authors 2022.
Conflict of interest statement
Conflict of interest: T.J. Corte reports grants, personal fees and nonfinancial support from Boehringer Ingelheim and Hoffman La Rochel grants and personal fees from Bristol Myers Squibb; grants from Avalyn Pharma and Biogen; and personal fees from Promedior, outside the submitted work. A.K.Y. Teoh reports conference fees from Boehringer Ingelheim and speaker fees from Roche outside the submitted work. The other co-authors declare no competing interests.
Figures
References
Publication types
LinkOut - more resources
Full Text Sources