

Molecular and Clinical Repercussions of GABA Transporter 1 Variants Gone Amiss: Links to Epilepsy and Developmental Spectrum Disorders
- PMID: 35295842
- PMCID: PMC7612498
- DOI: 10.3389/fmolb.2022.834498
Molecular and Clinical Repercussions of GABA Transporter 1 Variants Gone Amiss: Links to Epilepsy and Developmental Spectrum Disorders
Abstract
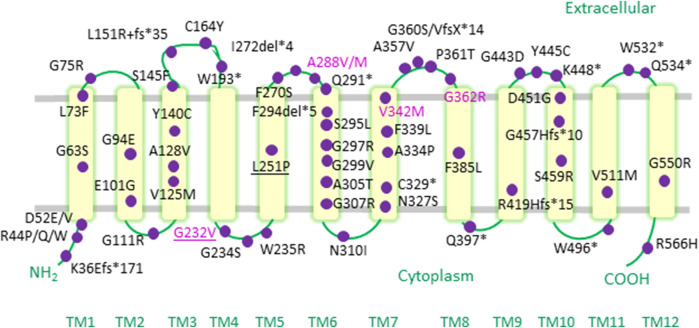
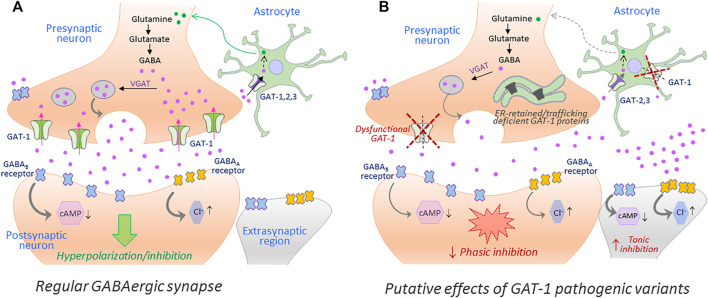
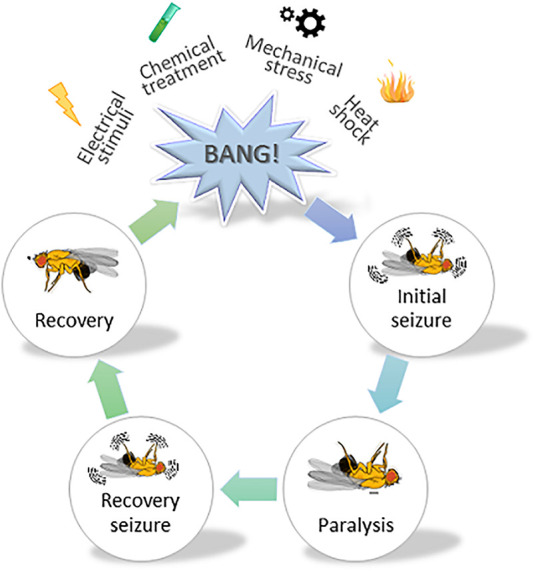
The human γ-aminobutyric acid (GABA) transporter 1 (hGAT-1) is the first member of the solute carrier 6 (SLC6) protein superfamily. GAT-1 (SLC6A1) is one of the main GABA transporters in the central nervous system. Its principal physiological role is retrieving GABA from the synapse into neurons and astrocytes, thus swiftly terminating neurotransmission. GABA is a key inhibitory neurotransmitter and shifts in GABAergic signaling can lead to pathological conditions, from anxiety and epileptic seizures to schizophrenia. Point mutations in the SLC6A1 gene frequently give rise to epilepsy, intellectual disability or autism spectrum disorders in the afflicted individuals. The mechanistic routes underlying these are still fairly unclear. Some loss-of-function variants impair the folding and intracellular trafficking of the protein (thus retaining the transporter in the endoplasmic reticulum compartment), whereas others, despite managing to reach their bona fide site of action at the cell surface, nonetheless abolish GABA transport activity (plausibly owing to structural/conformational defects). Whatever the molecular culprit(s), the physiological aftermath transpires into the absence of functional transporters, which in turn perturbs GABAergic actions. Dozens of mutations in the kin SLC6 family members are known to exhort protein misfolding. Such events typically elicit severe ailments in people, e.g., infantile parkinsonism-dystonia or X-linked intellectual disability, in the case of dopamine and creatine transporters, respectively. Flaws in protein folding can be rectified by small molecules known as pharmacological and/or chemical chaperones. The search for such apt remedies calls for a systematic investigation and categorization of the numerous disease-linked variants, by biochemical and pharmacological means in vitro (in cell lines and primary neuronal cultures) and in vivo (in animal models). We here give special emphasis to the utilization of the fruit fly Drosophila melanogaster as a versatile model in GAT-1-related studies. Jointly, these approaches can portray indispensable insights into the molecular factors underlying epilepsy, and ultimately pave the way for contriving efficacious therapeutic options for patients harboring pathogenic mutations in hGAT-1.
Keywords: Drosophila melanogaster; GABA transporter 1; autism; epilepsy; gamma-aminobutyric acid (GABA); intellectual disability; protein folding; transporter disease variants.
Copyright © 2022 Fischer, Kasture, Hummel and Sucic.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
References
-
- Asjad H. M. M., Kasture A., El-Kasaby A., Sackel M., Hummel T., Freissmuth M., et al. (2017). Pharmacochaperoning in a Drosophila Model System Rescues Human Dopamine Transporter Variants Associated with Infantile/juvenile Parkinsonism. J. Biol. Chem. 292, 19250–19265. 10.1074/JBC.M117.797092 - DOI - PMC - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases