

Lipid-storage myopathy with glycogen storage disease gene mutations mimicking polymyositis: a case report and review of the literature
- PMID: 35296144
- PMCID: PMC8943314
- DOI: 10.1177/03000605221084873
Lipid-storage myopathy with glycogen storage disease gene mutations mimicking polymyositis: a case report and review of the literature
Abstract
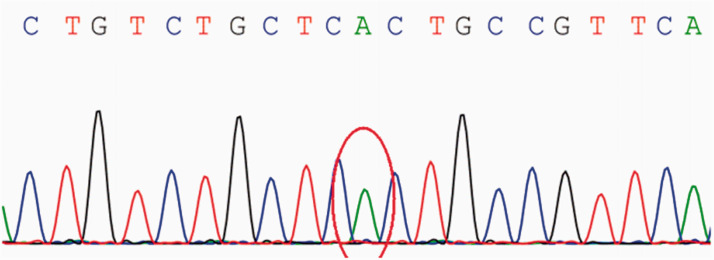
A 26-year-old Asian woman with persistent muscle weakness was diagnosed with polymyositis based on biopsy findings at another hospital 11 years ago. However, her symptoms fluctuated repeatedly under treatment with prednisone and immunosuppressive agents, and worsened 2 months prior to the current presentation. A second muscle biopsy suggested metabolic myopathy, and genetic testing revealed a novel c.1074C > T variant in the glycogen synthase 1 gene (GYS1), which is implicated in muscle glycogen storage disease type 0. However, no abnormalities in glycogen deposition were found by biopsy; rather, muscle fibers exhibited large intracellular lipid droplets. Furthermore, muscle strength was greatly restored and circulating levels of creatine kinase indicative of muscle degeneration greatly reduced by vitamin B2 treatment. Therefore, the final diagnosis was lipid storage myopathy.
Keywords: GYS1; Lipid storage myopathy; case report; glycogen storage disease type 0; polymyositis muscle; vitamin B2.
Conflict of interest statement
Figures



Similar articles
-
A myopathy with unusual features caused by PNPLA2 gene mutations.Muscle Nerve. 2015 Apr;51(4):609-13. doi: 10.1002/mus.24477. Epub 2015 Feb 28. Muscle Nerve. 2015. PMID: 25287355
-
A new phenotype of muscle glycogen synthase deficiency (GSD0B) characterized by an adult onset myopathy without cardiomyopathy.Neuromuscul Disord. 2022 Jul;32(7):582-589. doi: 10.1016/j.nmd.2022.03.008. Epub 2022 Mar 31. Neuromuscul Disord. 2022. PMID: 35641353
-
A novel PNPLA2 mutation causes neutral lipid storage disease with myopathy (NLSDM) presenting muscular dystrophic features with lipid storage and rimmed vacuoles.Clin Neuropathol. 2010 Nov-Dec;29(6):351-6. doi: 10.5414/npp29351. Clin Neuropathol. 2010. PMID: 21073837
-
Myopathies.Neurosurgery. 1979 Dec;5(6):747-58. doi: 10.1227/00006123-197912000-00017. Neurosurgery. 1979. PMID: 392333 Review.
-
Glycogen storage myopathies.Neurol Clin. 2000 Feb;18(1):125-50. doi: 10.1016/s0733-8619(05)70181-x. Neurol Clin. 2000. PMID: 10658171 Review.
Cited by
-
Proximal myopathy: causes and associated conditions.Discoveries (Craiova). 2022 Dec 31;10(4):e160. doi: 10.15190/d.2022.19. eCollection 2022 Oct-Dec. Discoveries (Craiova). 2022. PMID: 37483534 Free PMC article. Review.
-
A novel ETFDH mutation identified in a patient with riboflavin-responsive multiple acyl-CoA dehydrogenase deficiency.Intractable Rare Dis Res. 2025 Feb 28;14(1):76-80. doi: 10.5582/irdr.2024.01073. Intractable Rare Dis Res. 2025. PMID: 40046021 Free PMC article.
References
-
- Gagnier JJ, Kienle G, Altman DG, et al.; CARE Group. The CARE guidelines: consensus-based clinical case reporting guideline development. Headache 2013; 53: 1541–1547. - PubMed
-
- Wang Z, Hong D, Zhang W, et al.. Severe sensory neuropathy in patients with adult-onset multiple acyl-CoA dehydrogenase deficiency. Neuromuscul Disord 2016; 26: 170–175. DOI: 10.1016/j.nmd.2015.12.002. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
LinkOut - more resources
Full Text Sources
Medical
