

Identification and characterisation of spontaneous mutations causing deafness from a targeted knockout programme
- PMID: 35296311
- PMCID: PMC8928630
- DOI: 10.1186/s12915-022-01257-8
Identification and characterisation of spontaneous mutations causing deafness from a targeted knockout programme
Abstract
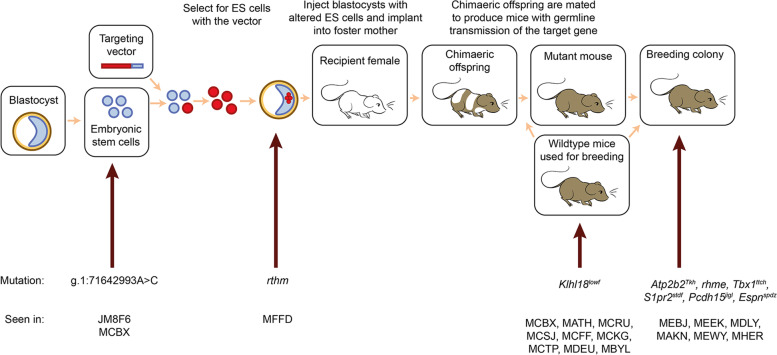
Background: Mice carrying targeted mutations are important for investigating gene function and the role of genes in disease, but off-target mutagenic effects associated with the processes of generating targeted alleles, for instance using Crispr, and culturing embryonic stem cells, offer opportunities for spontaneous mutations to arise. Identifying spontaneous mutations relies on the detection of phenotypes segregating independently of targeted alleles, and having a broad estimate of the level of mutations generated by intensive breeding programmes is difficult given that many phenotypes are easy to miss if not specifically looked for. Here we present data from a large, targeted knockout programme in which mice were analysed through a phenotyping pipeline. Such spontaneous mutations segregating within mutant lines may confound phenotypic analyses, highlighting the importance of record-keeping and maintaining correct pedigrees.
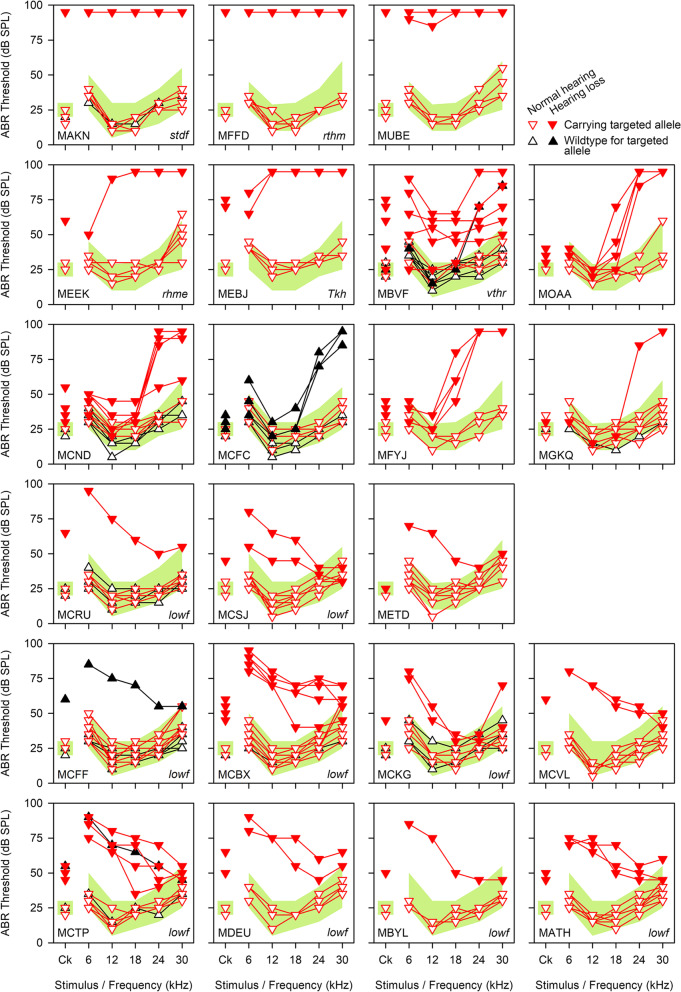
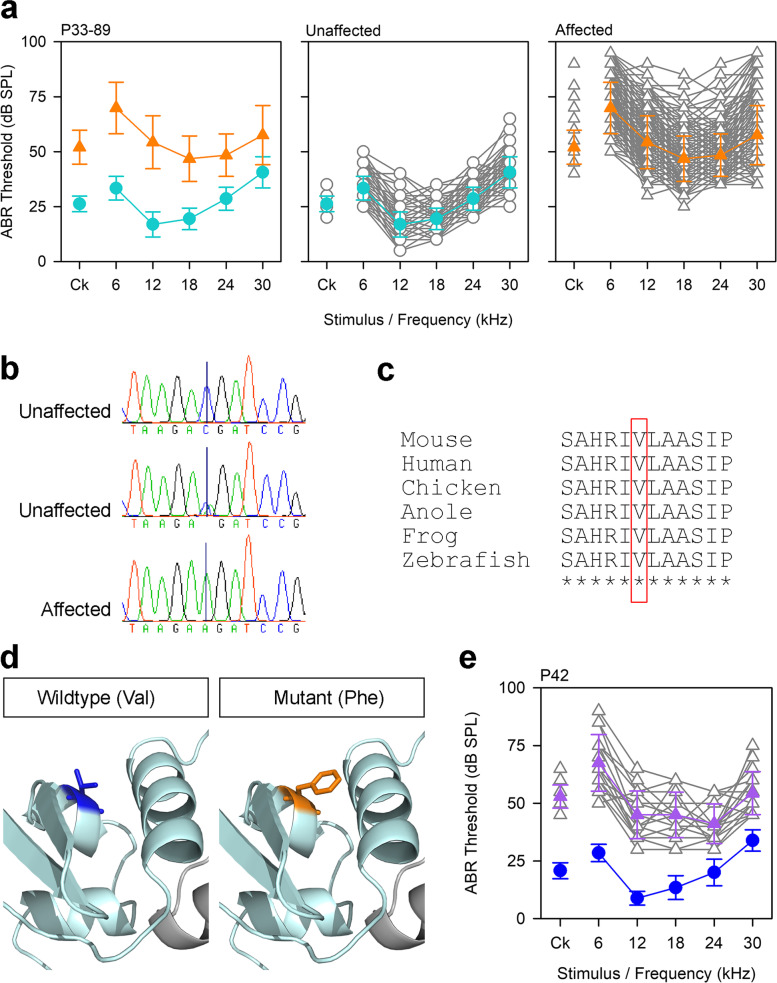
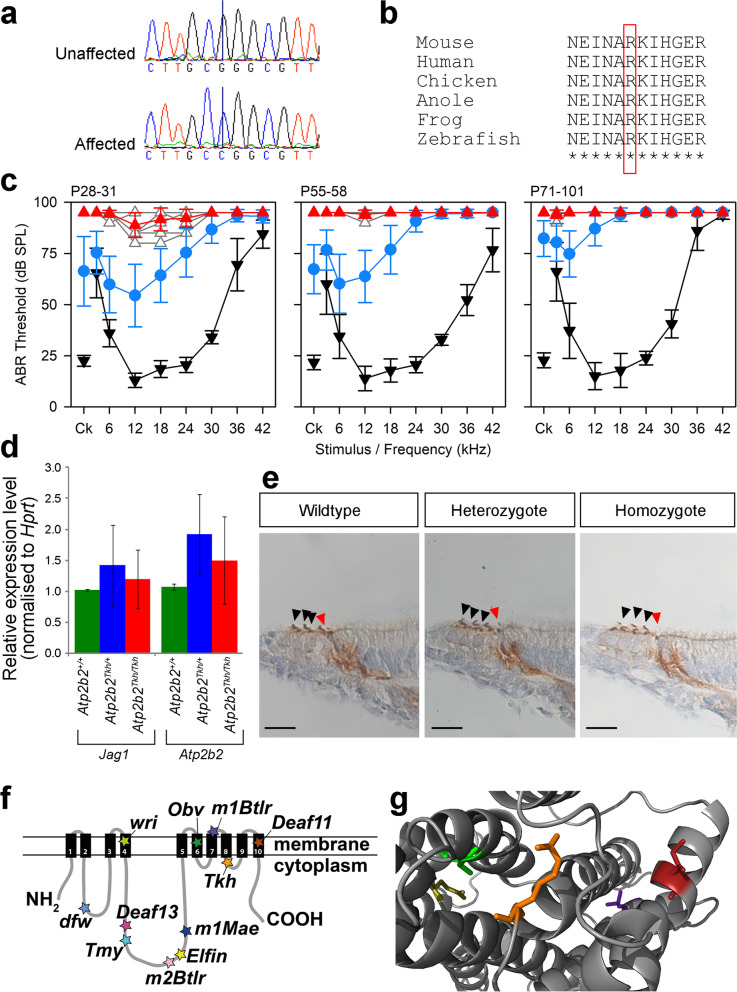
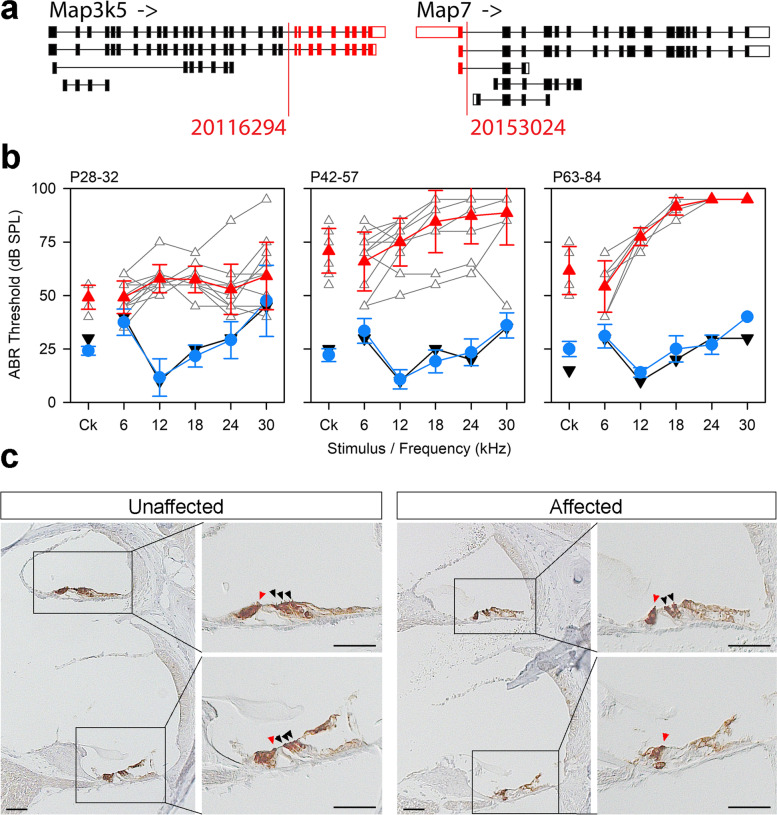
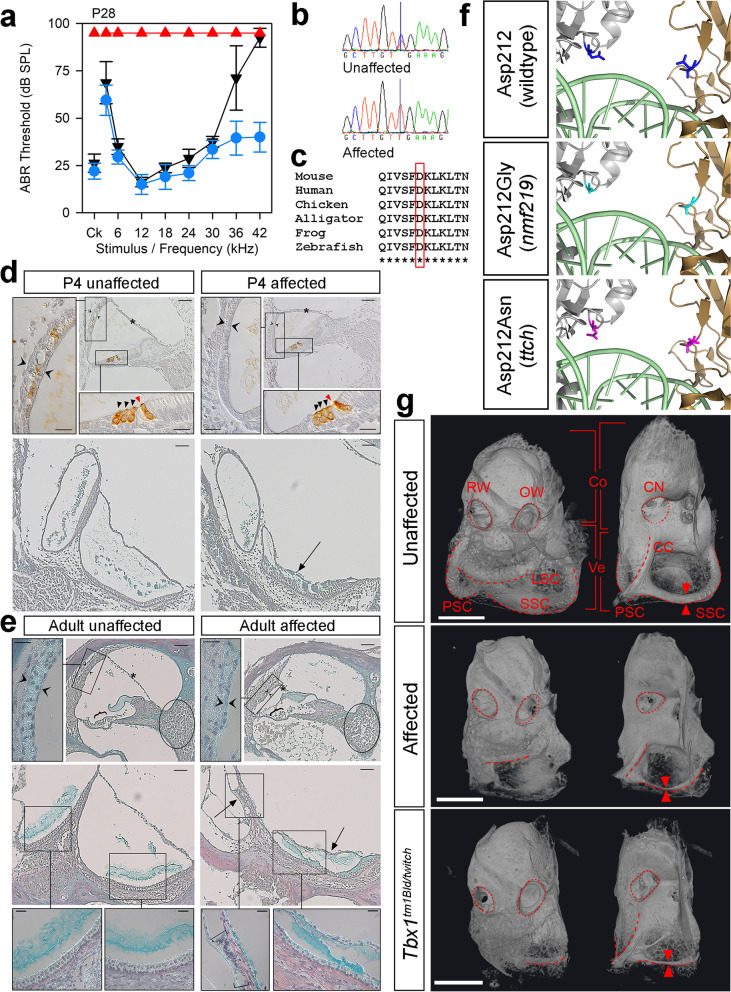
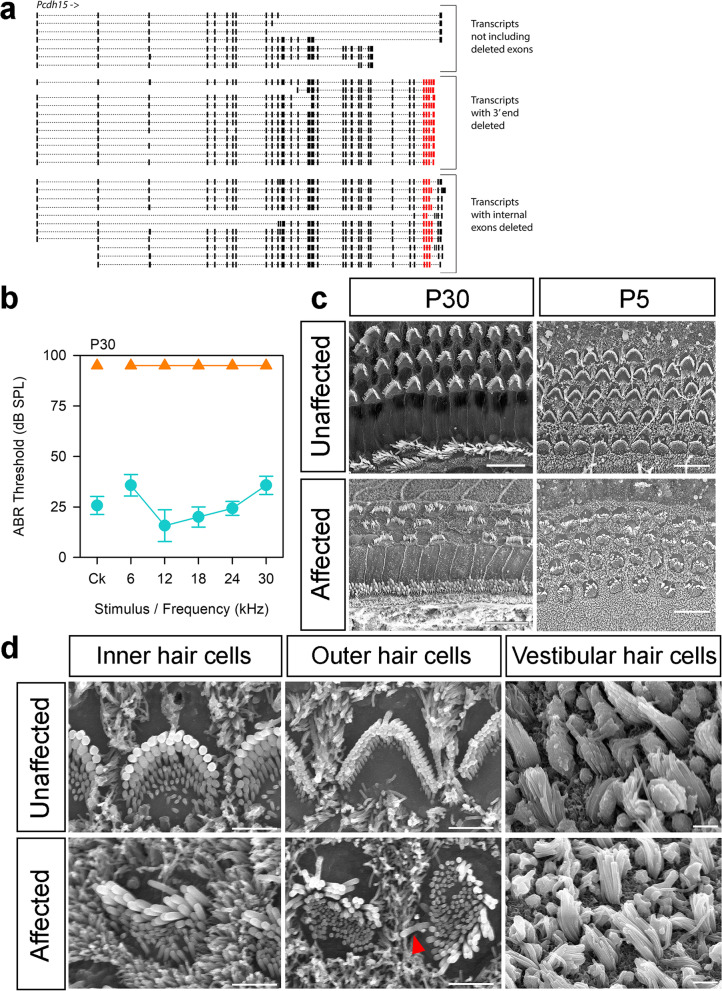
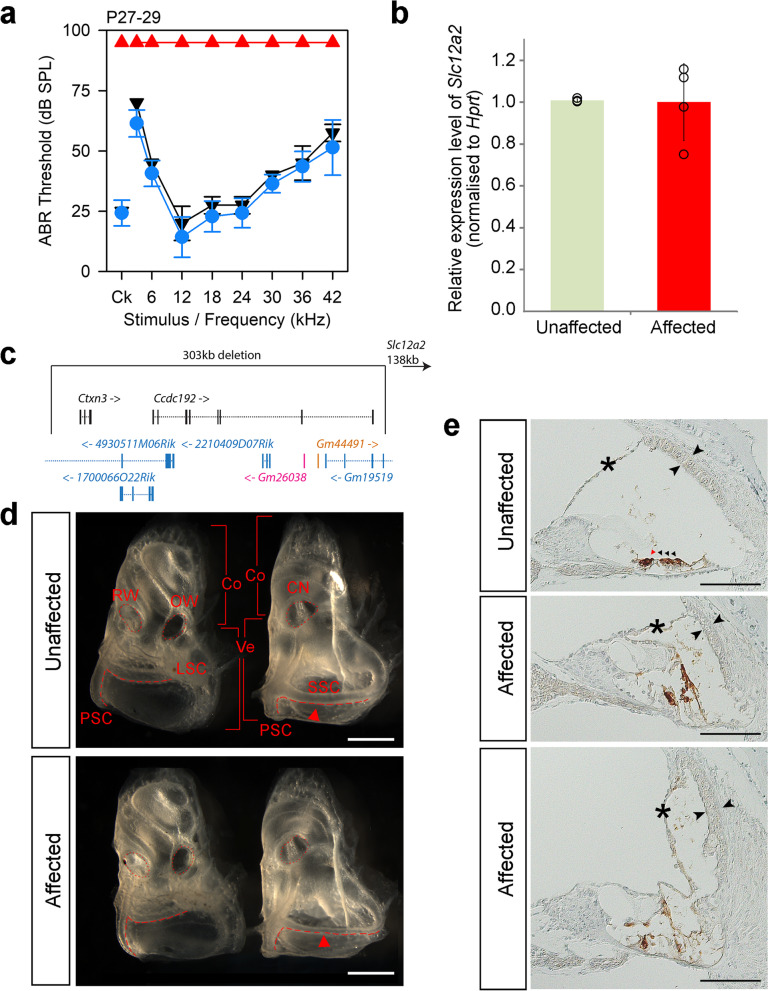
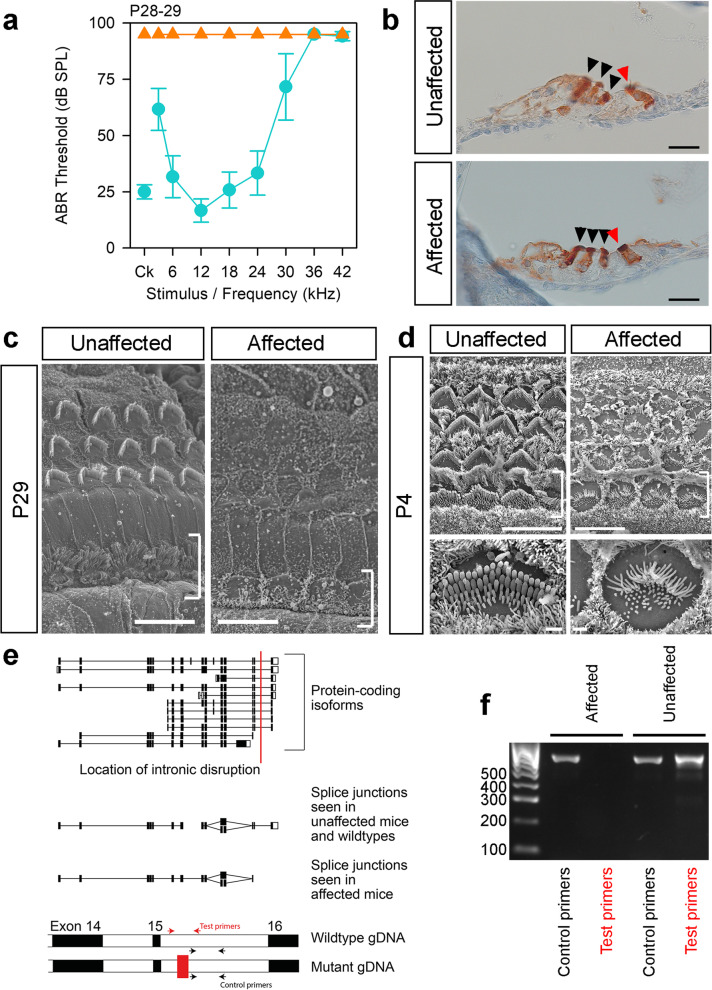
Results: Twenty-five lines out of 1311 displayed different deafness phenotypes that did not segregate with the targeted allele. We observed a variety of phenotypes by Auditory Brainstem Response (ABR) and behavioural assessment and isolated eight lines showing early-onset severe progressive hearing loss, later-onset progressive hearing loss, low frequency hearing loss, or complete deafness, with vestibular dysfunction. The causative mutations identified include deletions, insertions, and point mutations, some of which involve new genes not previously associated with deafness while others are new alleles of genes known to underlie hearing loss. Two of the latter show a phenotype much reduced in severity compared to other mutant alleles of the same gene. We investigated the ES cells from which these lines were derived and determined that only one of the 8 mutations could have arisen in the ES cell, and in that case, only after targeting. Instead, most of the non-segregating mutations appear to have occurred during breeding of mutant mice. In one case, the mutation arose within the wildtype colony used for expanding mutant lines.
Conclusions: Our data show that spontaneous mutations with observable effects on phenotype are a common side effect of intensive breeding programmes, including those underlying targeted mutation programmes. Such spontaneous mutations segregating within mutant lines may confound phenotypic analyses, highlighting the importance of record-keeping and maintaining correct pedigrees.
Keywords: Deafness; Large-scale mutagenesis programme; Non-segregating phenotypes; Progressive hearing loss; Spontaneous mutations.
© 2022. The Author(s).
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
References
-
- Willi M, Smith HE, Wang C, Liu C, Hennighausen L. Mutation frequency is not increased in CRISPR-Cas9-edited mice. Nat Methods. 2018;15(10):756–758. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
