

Novel CAPN1 missense variants in complex hereditary spastic paraplegia with early-onset psychosis
- PMID: 35297214
- PMCID: PMC8994985
- DOI: 10.1002/acn3.51531
Novel CAPN1 missense variants in complex hereditary spastic paraplegia with early-onset psychosis
Abstract
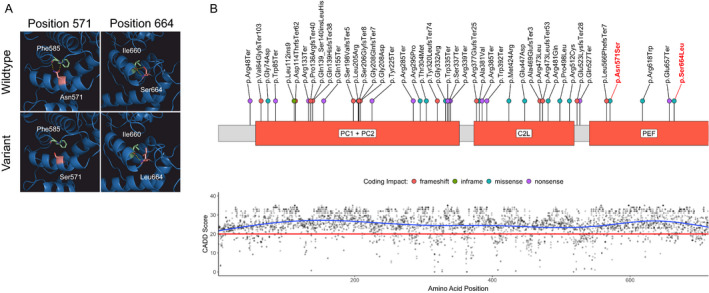
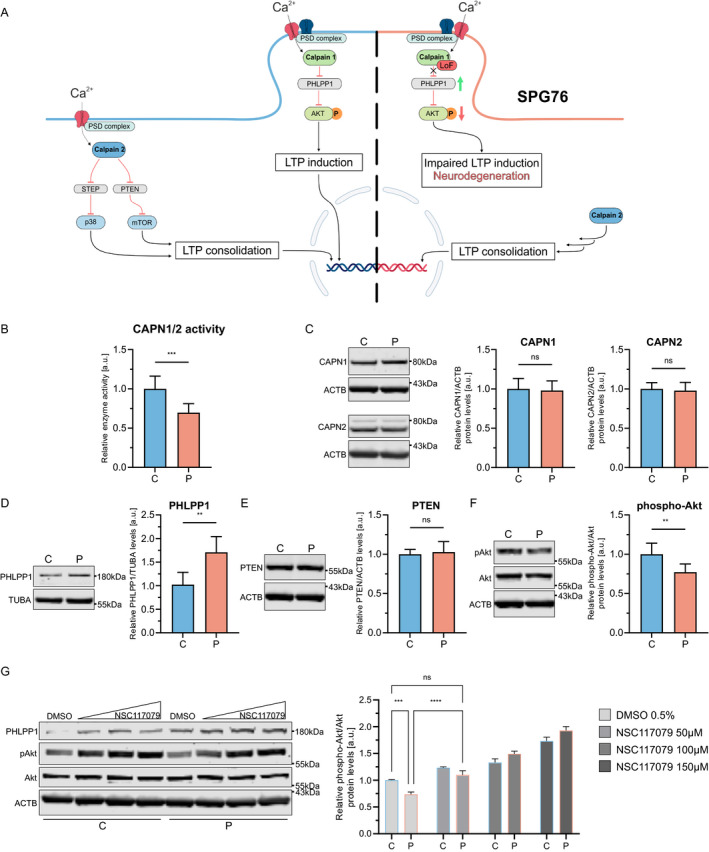
CAPN1-associated hereditary spastic paraplegia (SPG76) is a rare and clinically heterogenous syndrome due to loss of calpain-1 function. Here we illustrate a translational approach to the case of an 18-year-old patient who first presented with psychiatric symptoms followed by spastic gait, intention tremor, and neurogenic bladder dysfunction, consistent with a complex form of HSP. Exome sequencing showed compound-heterozygous missense variants in CAPN1 (NM_001198868.2: c.1712A>G (p.Asn571Ser)/c.1991C>T (p.Ser664Leu)) and a previously reported heterozygous stop-gain variant in RCL1. In silico analyses of the CAPN1 variants predicted a deleterious effect and in vitro functional studies confirmed reduced calpain-1 activity and dysregulated downstream signaling. These findings support a diagnosis of SPG76 and highlight that the psychiatric symptoms can precede the motor symptoms in HSP. Our results also suggest that multiple genes can potentially contribute to complex neuropsychiatric diseases.
© 2022 The Authors. Annals of Clinical and Translational Neurology published by Wiley Periodicals LLC on behalf of American Neurological Association.
Conflict of interest statement
D. E. F. received a speaker honorarium from the Movement Disorder Society, publishing royalties from the Cambridge University Press and reports research funding through a joint research agreement with Astellas Pharmaceuticals Inc.
Figures
References
-
- Shribman S, Reid E, Crosby AH, Houlden H, Warner TT. Hereditary spastic paraplegia: from diagnosis to emerging therapeutic approaches. Lancet Neurol. 2019;18(12):1136‐1146. - PubMed
-
- Erfanian Omidvar M, Torkamandi S, Rezaei S, et al. Genotype‐phenotype associations in hereditary spastic paraplegia: a systematic review and meta‐analysis on 13,570 patients. J Neurol. 2021;268(6):2065‐2082. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
