

Multiple causal variants underlie genetic associations in humans
- PMID: 35298243
- PMCID: PMC9725108
- DOI: 10.1126/science.abj5117
Multiple causal variants underlie genetic associations in humans
Abstract
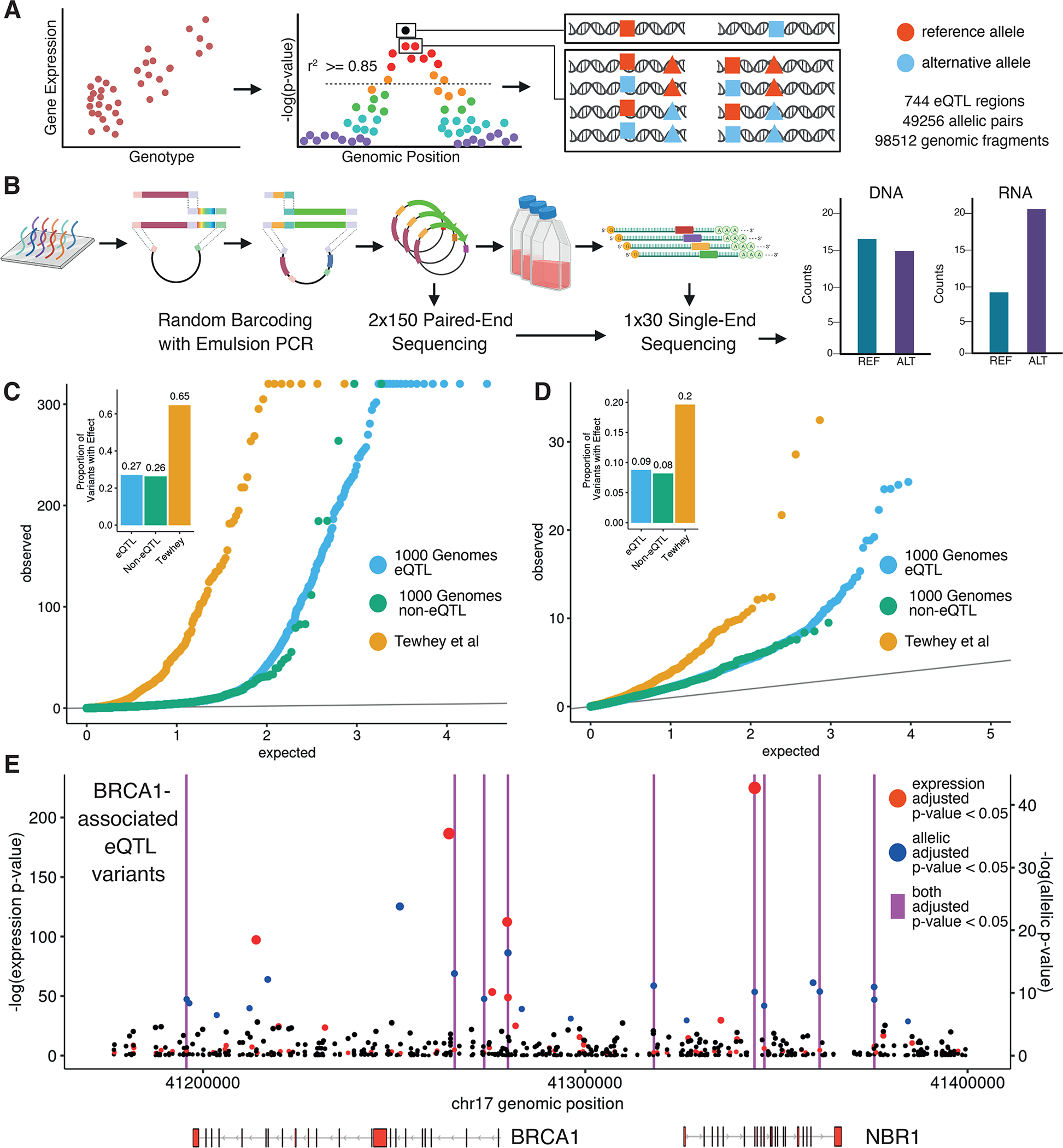
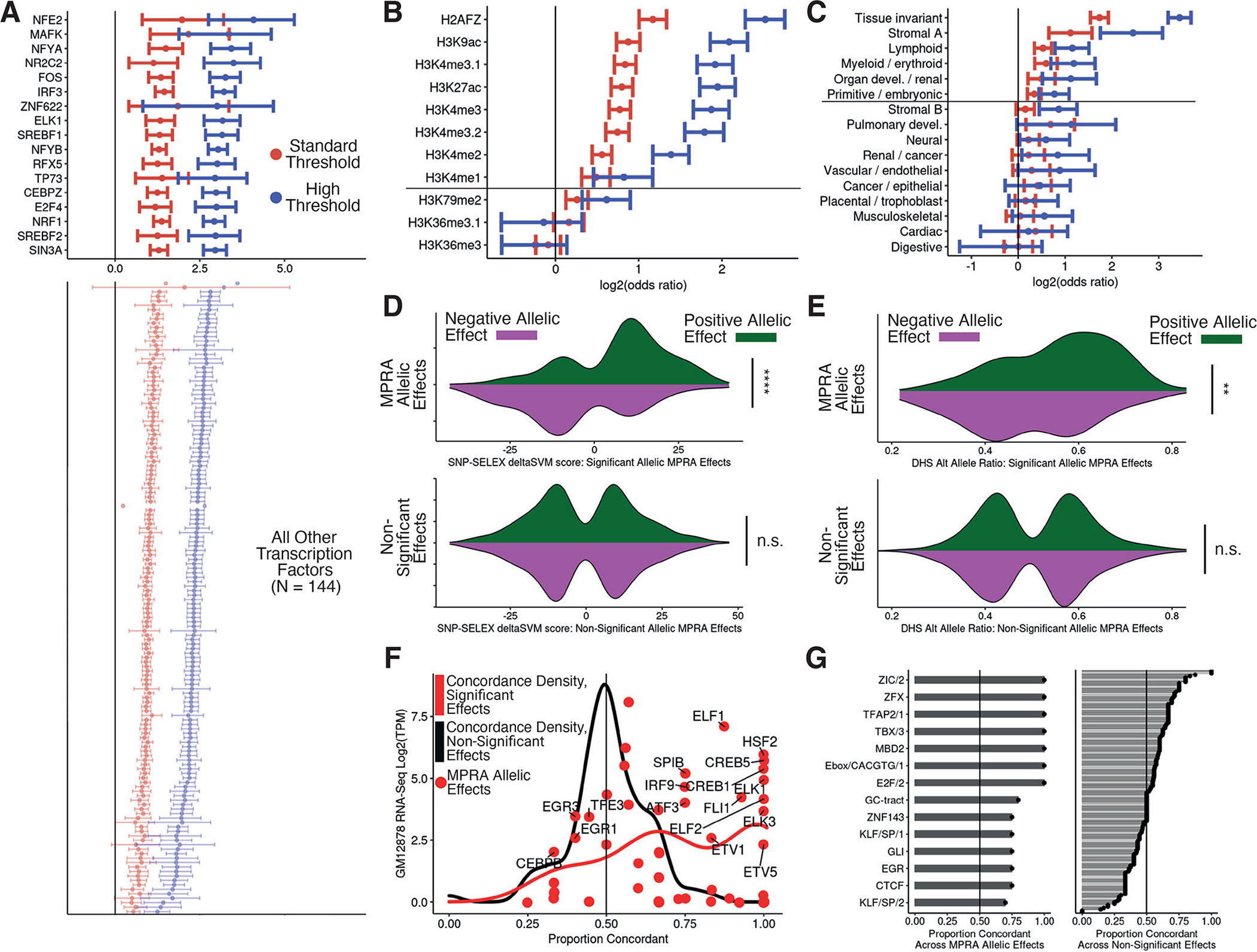
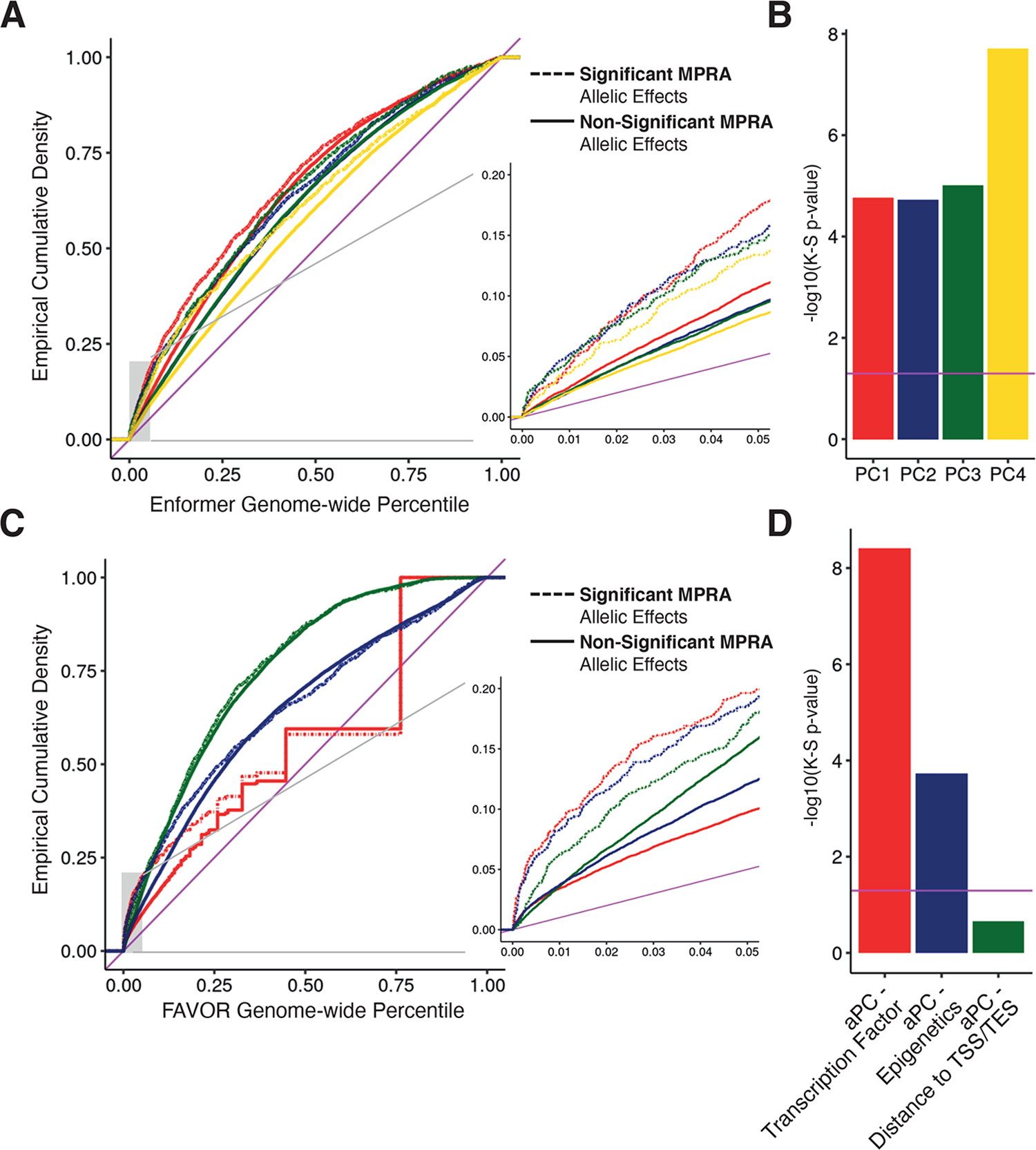
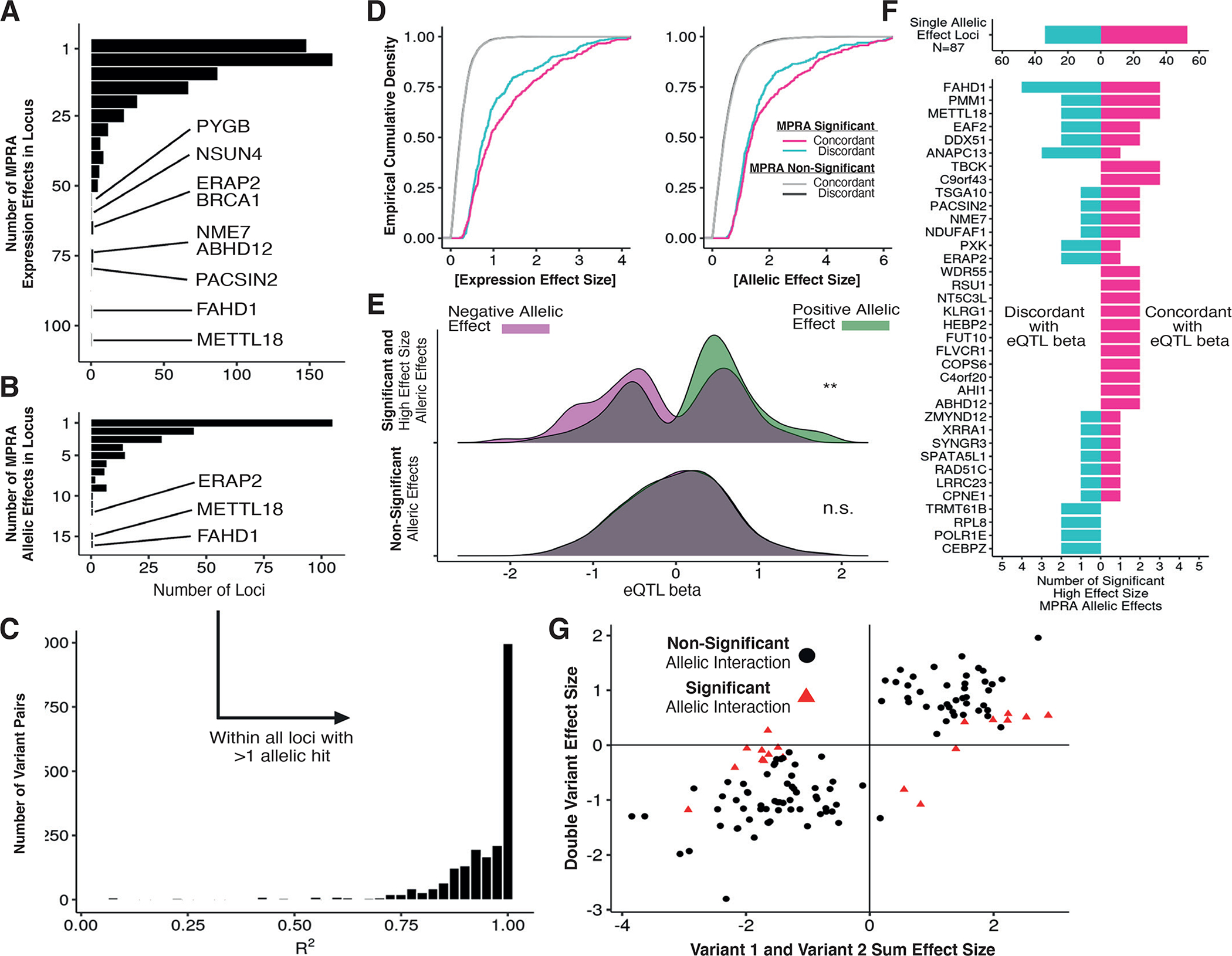
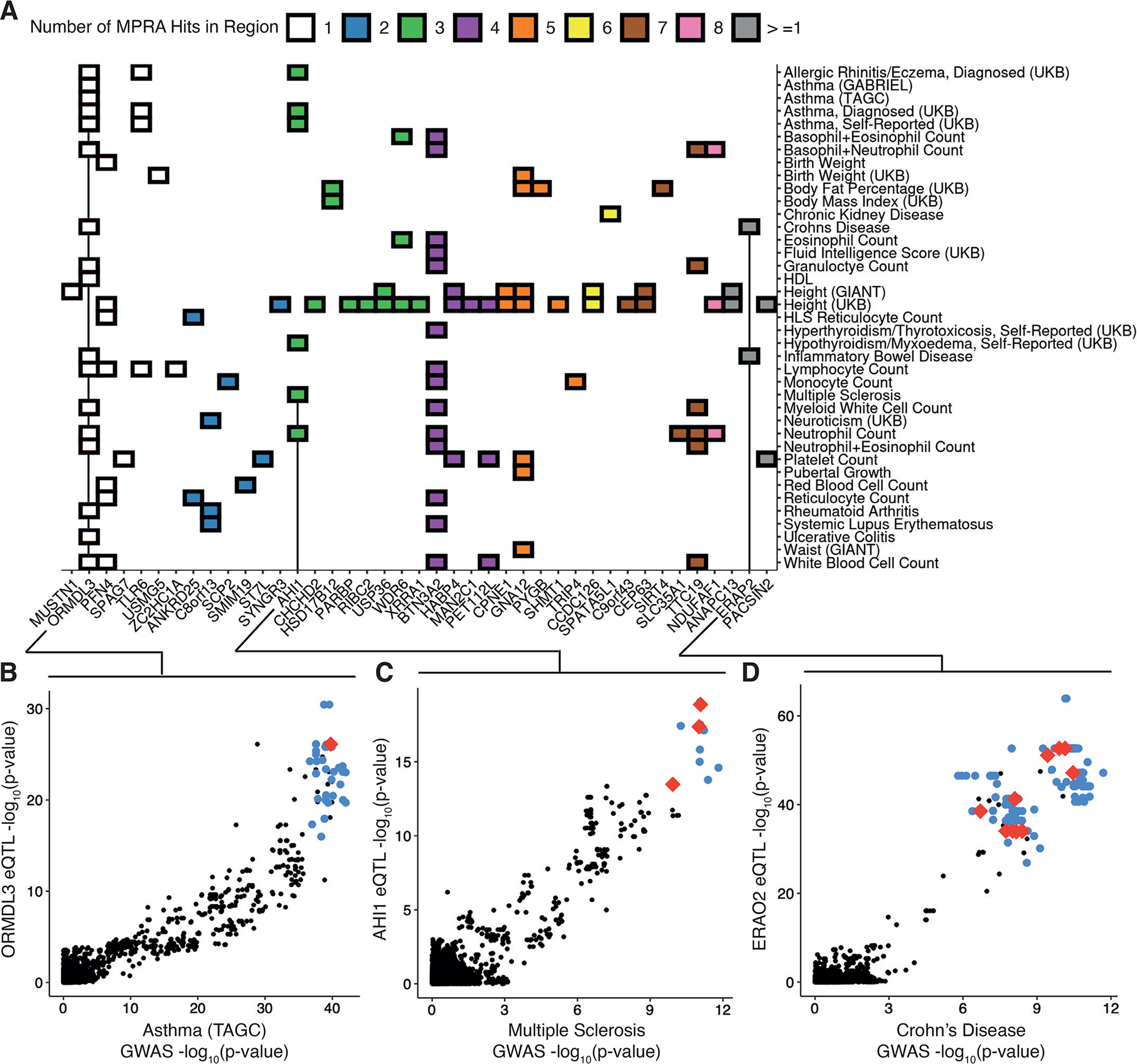
Associations between genetic variation and traits are often in noncoding regions with strong linkage disequilibrium (LD), where a single causal variant is assumed to underlie the association. We applied a massively parallel reporter assay (MPRA) to functionally evaluate genetic variants in high, local LD for independent cis-expression quantitative trait loci (eQTL). We found that 17.7% of eQTLs exhibit more than one major allelic effect in tight LD. The detected regulatory variants were highly and specifically enriched for activating chromatin structures and allelic transcription factor binding. Integration of MPRA profiles with eQTL/complex trait colocalizations across 114 human traits and diseases identified causal variant sets demonstrating how genetic association signals can manifest through multiple, tightly linked causal variants.
Conflict of interest statement
Figures
Comment in
-
Fine-mapping causal variants - why finding 'the one' can be futile.Nat Rev Genet. 2022 May;23(5):261. doi: 10.1038/s41576-022-00484-7. Nat Rev Genet. 2022. PMID: 35351991 No abstract available.
References
-
- Buniello A, MacArthur JAL, Cerezo M, Harris LW, Hayhurst J, Malangone C, McMahon A, Morales J, Mountjoy E, Sollis E, Suveges D, Vrousgou O, Whetzel PL, Amode R, Guillen JA, Riat HS, Trevanion SJ, Hall P, Junkins H, Flicek P, Burdett T, Hindorff LA, Cunningham F, Parkinson H, The NHGRI-EBI GWAS Catalog of published genome-wide association studies, targeted arrays and summary statistics 2019. Nucleic Acids Res. 47, D1005–D1012 (2019). - PMC - PubMed
-
- Tam V, Patel N, Turcotte M, Bossé Y, Paré G, Meyre D, Benefits and limitations of genome-wide association studies. Nat. Rev. Genet. 20, 467–484 (2019). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
