

Post-Traumatic Epilepsy and Comorbidities: Advanced Models, Molecular Mechanisms, Biomarkers, and Novel Therapeutic Interventions
- PMID: 35302046
- PMCID: PMC8973512
- DOI: 10.1124/pharmrev.121.000375
Post-Traumatic Epilepsy and Comorbidities: Advanced Models, Molecular Mechanisms, Biomarkers, and Novel Therapeutic Interventions
Abstract
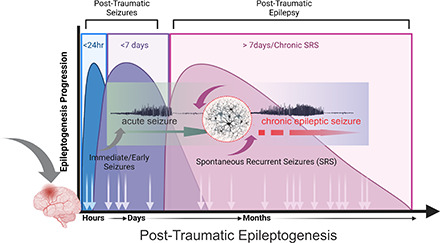
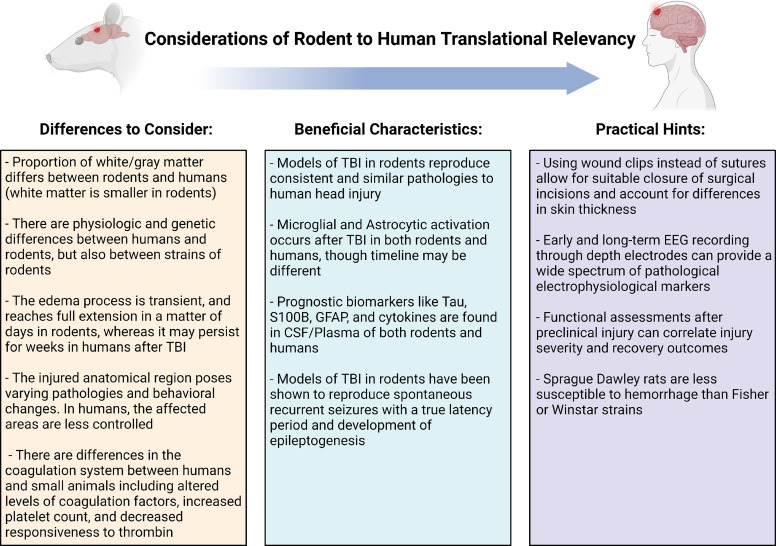
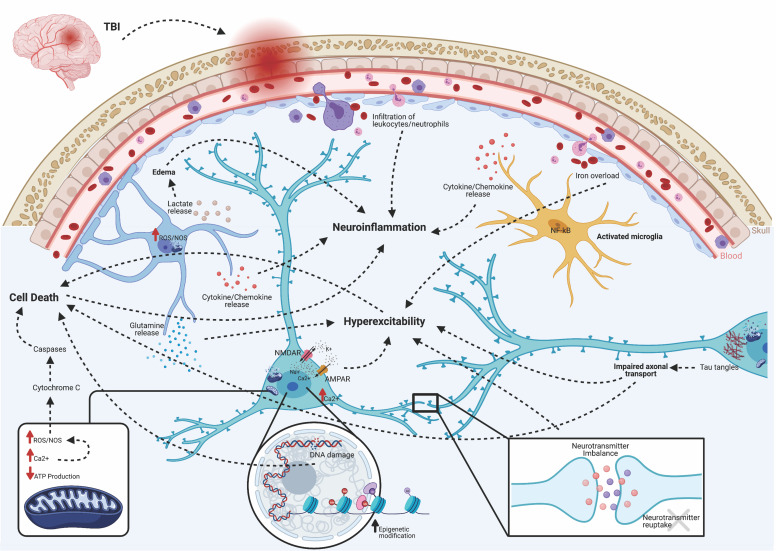
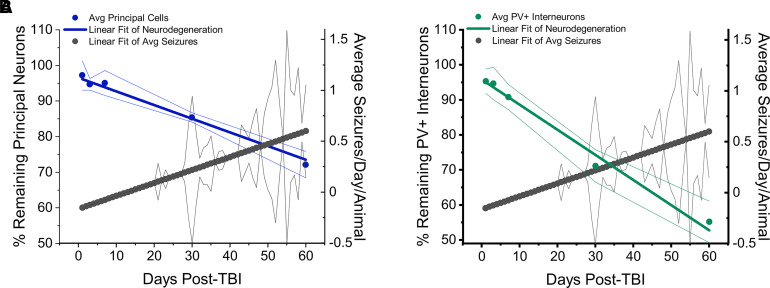
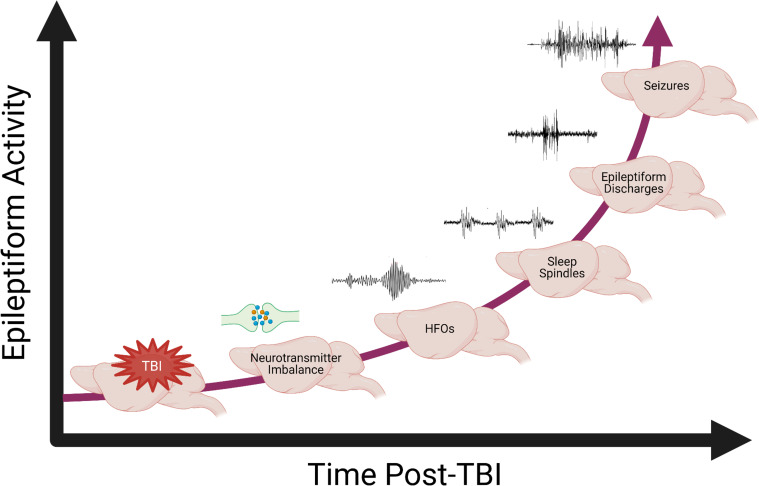
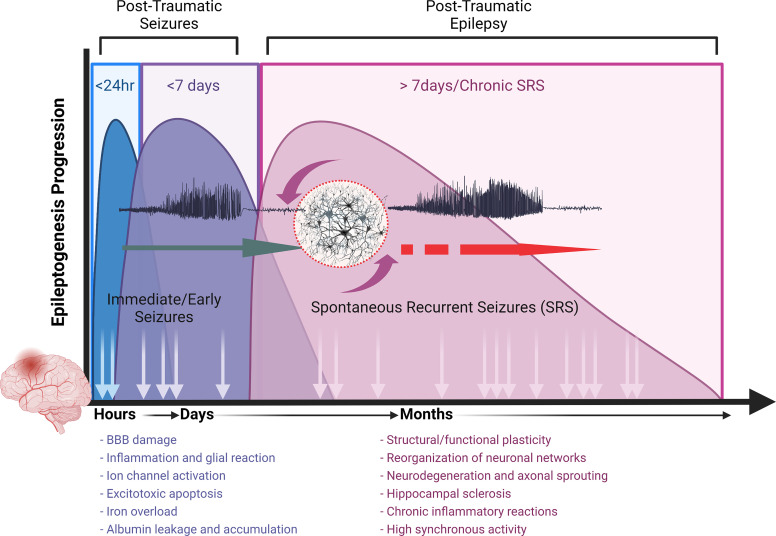
Post-traumatic epilepsy (PTE) is one of the most devastating long-term, network consequences of traumatic brain injury (TBI). There is currently no approved treatment that can prevent onset of spontaneous seizures associated with brain injury, and many cases of PTE are refractory to antiseizure medications. Post-traumatic epileptogenesis is an enduring process by which a normal brain exhibits hypersynchronous excitability after a head injury incident. Understanding the neural networks and molecular pathologies involved in epileptogenesis are key to preventing its development or modifying disease progression. In this article, we describe a critical appraisal of the current state of PTE research with an emphasis on experimental models, molecular mechanisms of post-traumatic epileptogenesis, potential biomarkers, and the burden of PTE-associated comorbidities. The goal of epilepsy research is to identify new therapeutic strategies that can prevent PTE development or interrupt the epileptogenic process and relieve associated neuropsychiatric comorbidities. Therefore, we also describe current preclinical and clinical data on the treatment of PTE sequelae. Differences in injury patterns, latency period, and biomarkers are outlined in the context of animal model validation, pathophysiology, seizure frequency, and behavior. Improving TBI recovery and preventing seizure onset are complex and challenging tasks; however, much progress has been made within this decade demonstrating disease modifying, anti-inflammatory, and neuroprotective strategies, suggesting this goal is pragmatic. Our understanding of PTE is continuously evolving, and improved preclinical models allow for accelerated testing of critically needed novel therapeutic interventions in military and civilian persons at high risk for PTE and its devastating comorbidities. SIGNIFICANCE STATEMENT: Post-traumatic epilepsy is a chronic seizure condition after brain injury. With few models and limited understanding of the underlying progression of epileptogenesis, progress is extremely slow to find a preventative treatment for PTE. This study reviews the current state of modeling, pathology, biomarkers, and potential interventions for PTE and comorbidities. There's new optimism in finding a drug therapy for preventing PTE in people at risk, such as after traumatic brain injury, concussion, and serious brain injuries, especially in military persons.
Copyright © 2022 by The Author(s).
Figures
References
-
- Abdelmalik PA, Boorman DW, Tracy J, Jallo J, Rincon F (2016) Acute traumatic coagulopathy accompanying isolated traumatic brain injury is associated with worse long-term functional and cognitive outcomes. Neurocrit Care 24:361–370. - PubMed
-
- Adeyemo BO, Biederman J, Zafonte R, Kagan E, Spencer TJ, Uchida M, Kenworthy T, Spencer AE, Faraone SV (2014) Mild traumatic brain injury and ADHD: a systematic review of the literature and meta-analysis. J Atten Disord 18:576–584. - PubMed
-
- Agoston DV, Shutes-David A, Peskind ER (2017) Biofluid biomarkers of traumatic brain injury. Brain Inj 31:1195–1203. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
