

Global Population Genomics of Two Subspecies of Cryptosporidium hominis during 500 Years of Evolution
- PMID: 35302613
- PMCID: PMC9004413
- DOI: 10.1093/molbev/msac056
Global Population Genomics of Two Subspecies of Cryptosporidium hominis during 500 Years of Evolution
Abstract
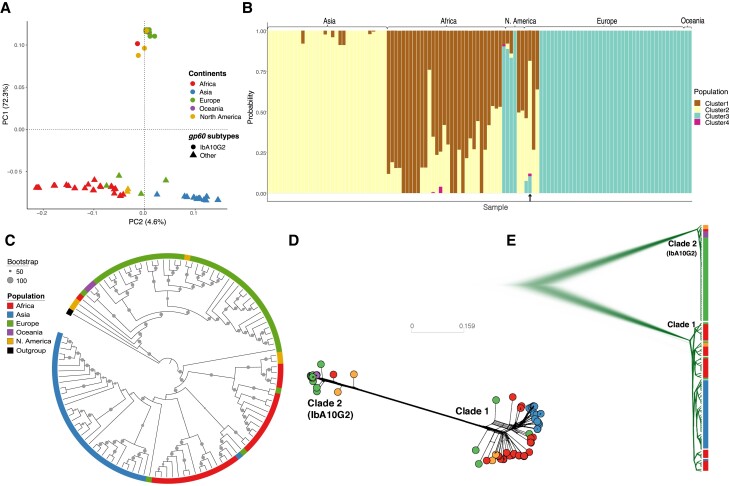
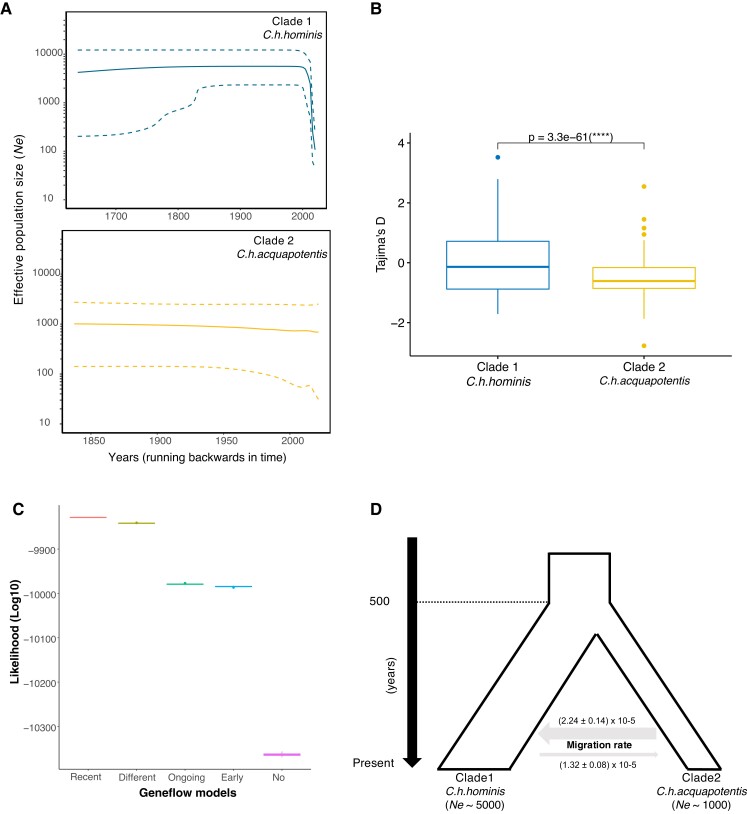
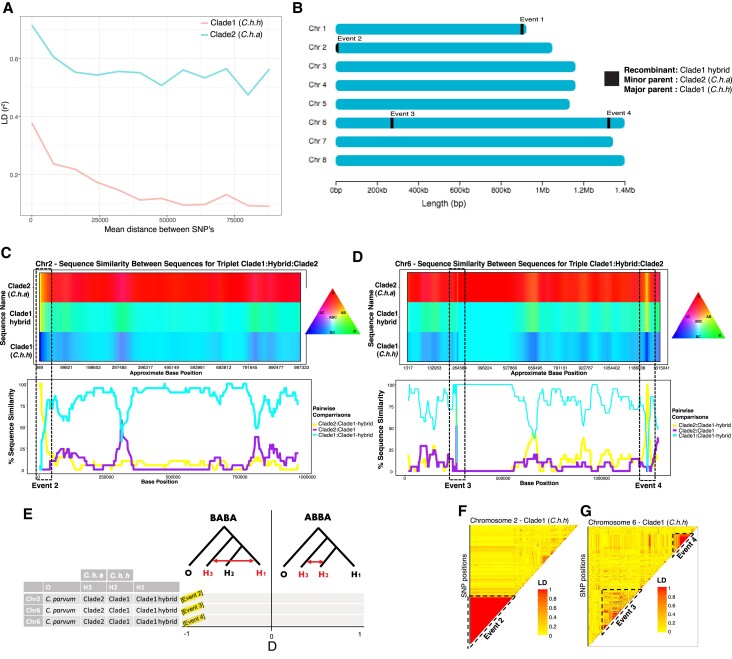
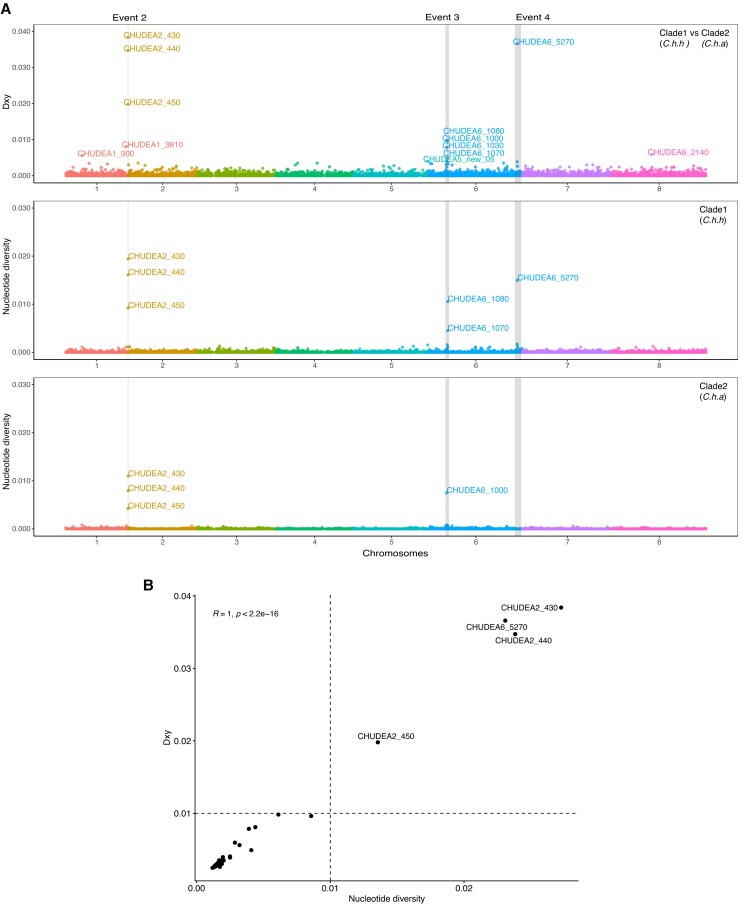
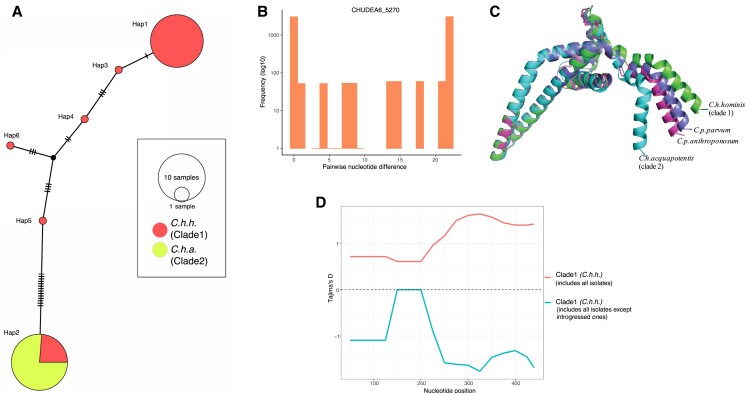
Cryptosporidiosis is a major global health problem and a primary cause of diarrhea, particularly in young children in low- and middle-income countries (LMICs). The zoonotic Cryptosporidium parvum and anthroponotic Cryptosporidium hominis cause most human infections. Here, we present a comprehensive whole-genome study of C. hominis, comprising 114 isolates from 16 countries within five continents. We detect two lineages with distinct biology and demography, which diverged circa 500 years ago. We consider these lineages two subspecies and propose the names C. hominis hominis and C. hominis aquapotentis (gp60 subtype IbA10G2). In our study, C. h. hominis is almost exclusively represented by isolates from LMICs in Africa and Asia and appears to have undergone recent population contraction. In contrast, C. h. aquapotentis was found in high-income countries, mainly in Europe, North America, and Oceania, and appears to be expanding. Notably, C. h. aquapotentis is associated with high rates of direct human-to-human transmission, which may explain its success in countries with well-developed environmental sanitation infrastructure. Intriguingly, we detected genomic regions of introgression following secondary contact between the subspecies. This resulted in high diversity and divergence in genomic islands of putative virulence genes, including muc5 (CHUDEA2_430) and a hypothetical protein (CHUDEA6_5270). This diversity is maintained by balancing selection, suggesting a co-evolutionary arms race with the host. Finally, we find that recent gene flow from C. h. aquapotentis to C. h. hominis, likely associated with increased human migration, maybe driving the evolution of more virulent C. hominis variants.
Keywords: Cryptosporidium hominis; comparative genomics; evolution; gene flow; population genetics; population structure; recombination; secondary contact; speciation; whole genome sequencing.
© The Author(s) 2022. Published by Oxford University Press on behalf of Society for Molecular Biology and Evolution.
Figures
References
-
- Abrahamsen MS, Templeton TJ, Enomoto S, Abrahante JE, Zhu G, Lancto CA, Deng M, Liu C, Widmer G, Tzipori S, et al. . 2004. Complete genome sequence of the apicomplexan, Cryptosporidium parvum. Science 304:441–445. - PubMed
-
- Bouckaert RR, Heled J. 2014. DensiTree 2: seeing trees through the forest. bioRxiv 012401.
