

The role of inflammation in neurodegeneration: novel insights into the role of the immune system in C9orf72 HRE-mediated ALS/FTD
- PMID: 35303907
- PMCID: PMC8932121
- DOI: 10.1186/s13024-022-00525-z
The role of inflammation in neurodegeneration: novel insights into the role of the immune system in C9orf72 HRE-mediated ALS/FTD
Abstract
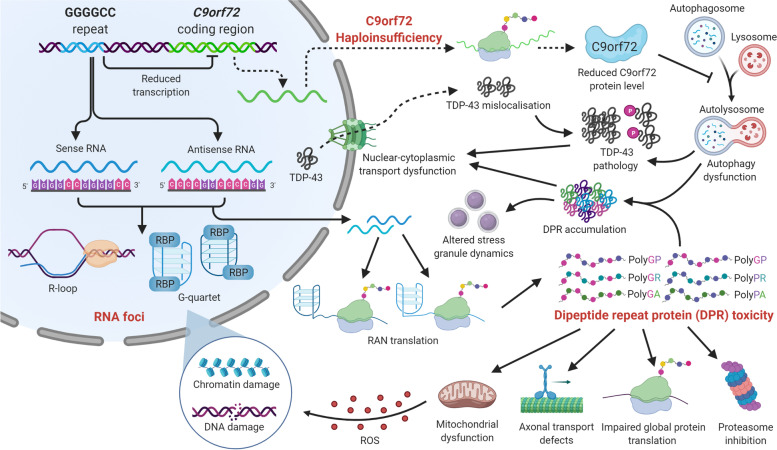
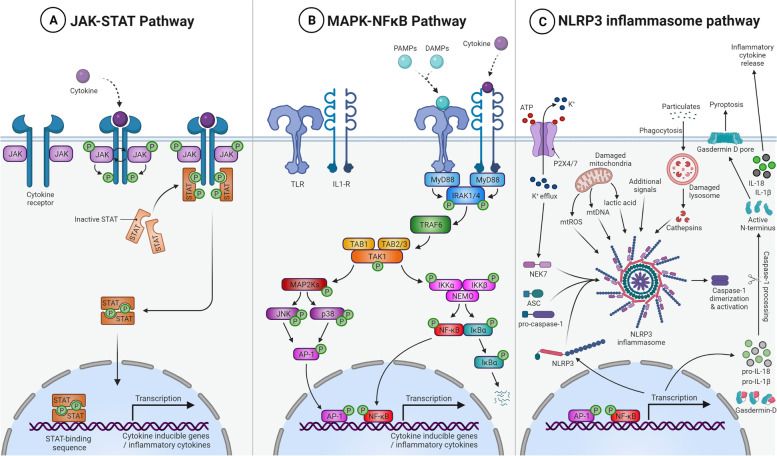
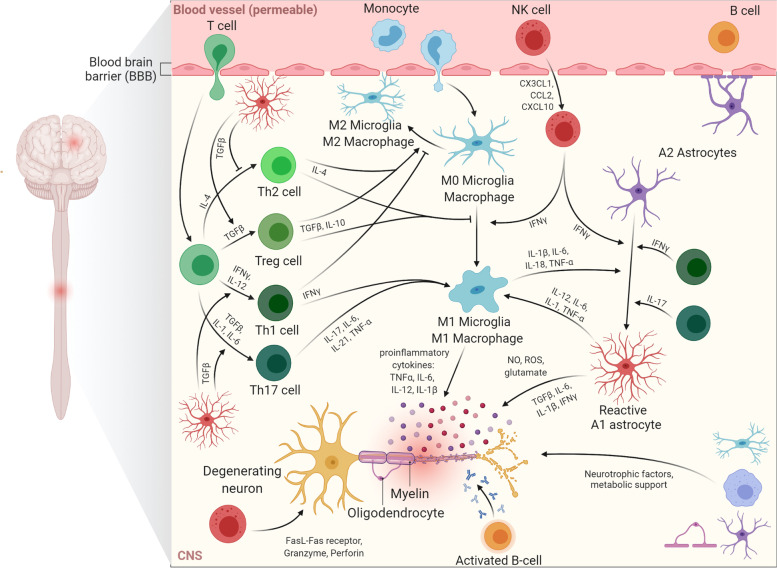
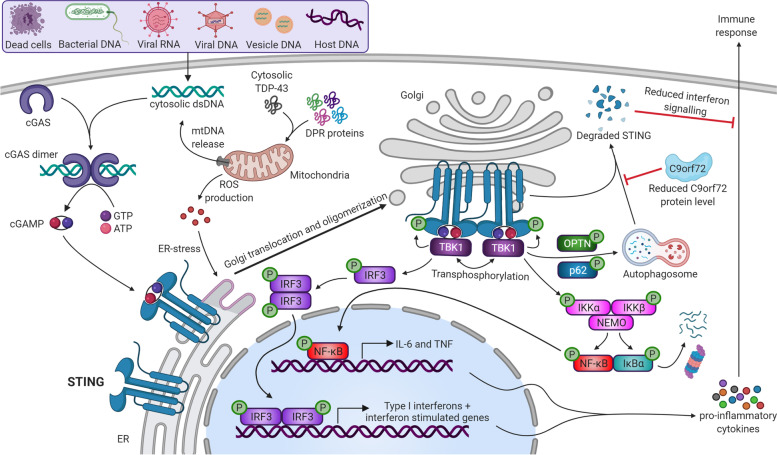
Neuroinflammation is an important hallmark of amyotrophic lateral sclerosis (ALS) and frontotemporal lobar degeneration (FTLD). An inflammatory reaction to neuronal injury is deemed vital for neuronal health and homeostasis. However, a continued activation of the inflammatory response can be detrimental to remaining neurons and aggravate the disease process. Apart from a disease modifying role, some evidence suggests that neuroinflammation may also contribute to the upstream cause of the disease. In this review, we will first focus on the role of neuroinflammation in the pathogenesis of chromosome 9 open reading frame 72 gene (C9orf72) hexanucleotide repeat expansions (HRE)-mediated ALS/FTD (C9-ALS/FTD). Additionally, we will discuss evidence from ex vivo and in vivo studies and finally, we briefly summarize the trials and progress of anti-inflammatory therapies.
Keywords: Anti-inflammatory therapies; Astrocytes; C9orf72 HRE-mediated ALS/FTD; In-vivo and ex-vivo models; Microglia; Neuroinflammation; Peripheral immune cells.
© 2022. The Author(s).
Conflict of interest statement
The authors declare to have no conflicts of interests.
Figures
References
-
- Lomen-Hoerth C, Anderson T, Miller B. The overlap of amyotrophic lateral sclerosis and frontotemporal dementia. Neurology. 2002;59(7):1077–1079. - PubMed
-
- Neumann M, Mackenzie IRA. Review: Neuropathology of non-tau frontotemporal lobar degeneration. Neuropathology and applied neurobiology. 2019;45(1):19–40. - PubMed
-
- Hardiman O, Al-Chalabi A, Chio A, Corr EM, Logroscino G, Robberecht W, et al. Amyotrophic lateral sclerosis. Nat Rev Dis Primers. 2017;5(3):17071. - PubMed
-
- Oskarsson B, Gendron TF, Staff NP. Amyotrophic Lateral Sclerosis: An Update for 2018. Mayo Clin Proc. 2018;93(11):1617–1628. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
