

The interplay of additivity, dominance, and epistasis on fitness in a diploid yeast cross
- PMID: 35304450
- PMCID: PMC8933436
- DOI: 10.1038/s41467-022-29111-z
The interplay of additivity, dominance, and epistasis on fitness in a diploid yeast cross
Abstract
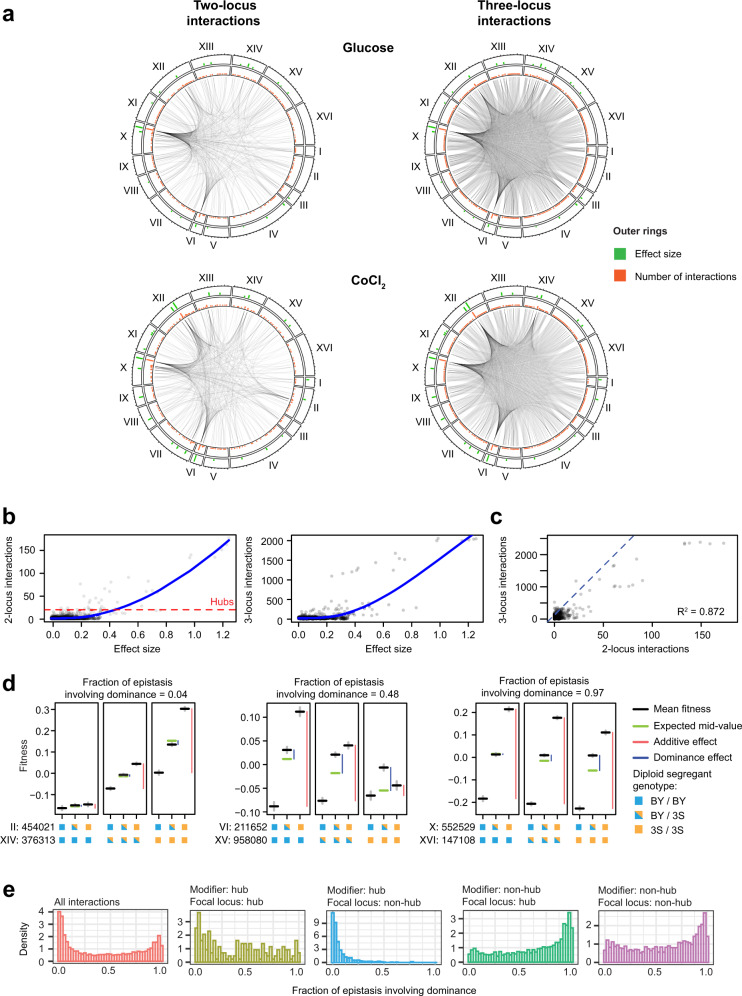
In diploid species, genetic loci can show additive, dominance, and epistatic effects. To characterize the contributions of these different types of genetic effects to heritable traits, we use a double barcoding system to generate and phenotype a panel of ~200,000 diploid yeast strains that can be partitioned into hundreds of interrelated families. This experiment enables the detection of thousands of epistatic loci, many whose effects vary across families. Here, we show traits are largely specified by a small number of hub loci with major additive and dominance effects, and pervasive epistasis. Genetic background commonly influences both the additive and dominance effects of loci, with multiple modifiers typically involved. The most prominent dominance modifier in our data is the mating locus, which has no effect on its own. Our findings show that the interplay between additivity, dominance, and epistasis underlies a complex genotype-to-phenotype map in diploids.
© 2022. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures




References
-
- Mackay TFC, Stone EA, Ayroles JF. The genetics of quantitative traits: challenges and prospects. Nat. Rev. Genet. 2009;10:565–577. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
