

Targeting CDK4 and CDK6 in cancer
- PMID: 35304604
- PMCID: PMC9149100
- DOI: 10.1038/s41568-022-00456-3
Targeting CDK4 and CDK6 in cancer
Abstract
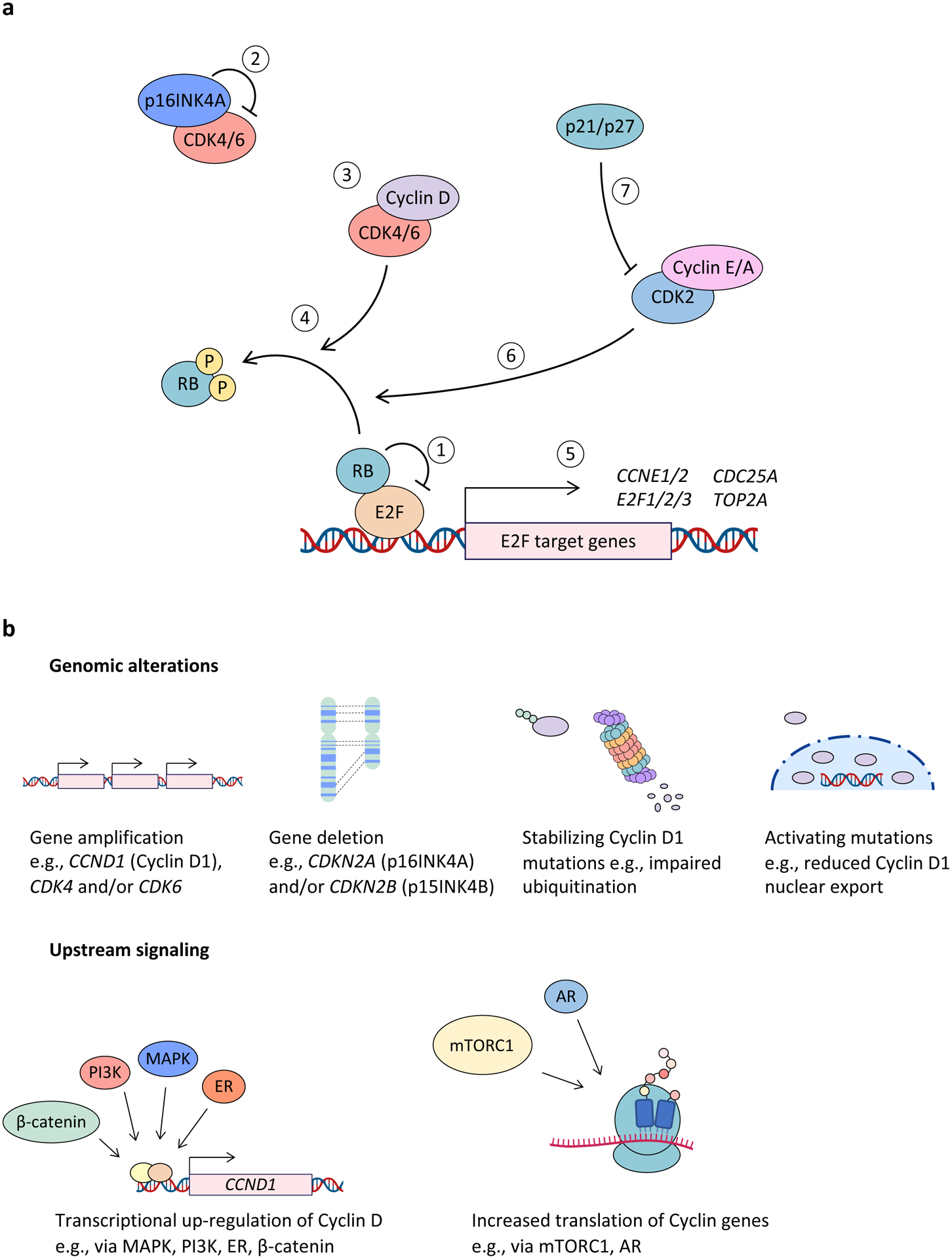
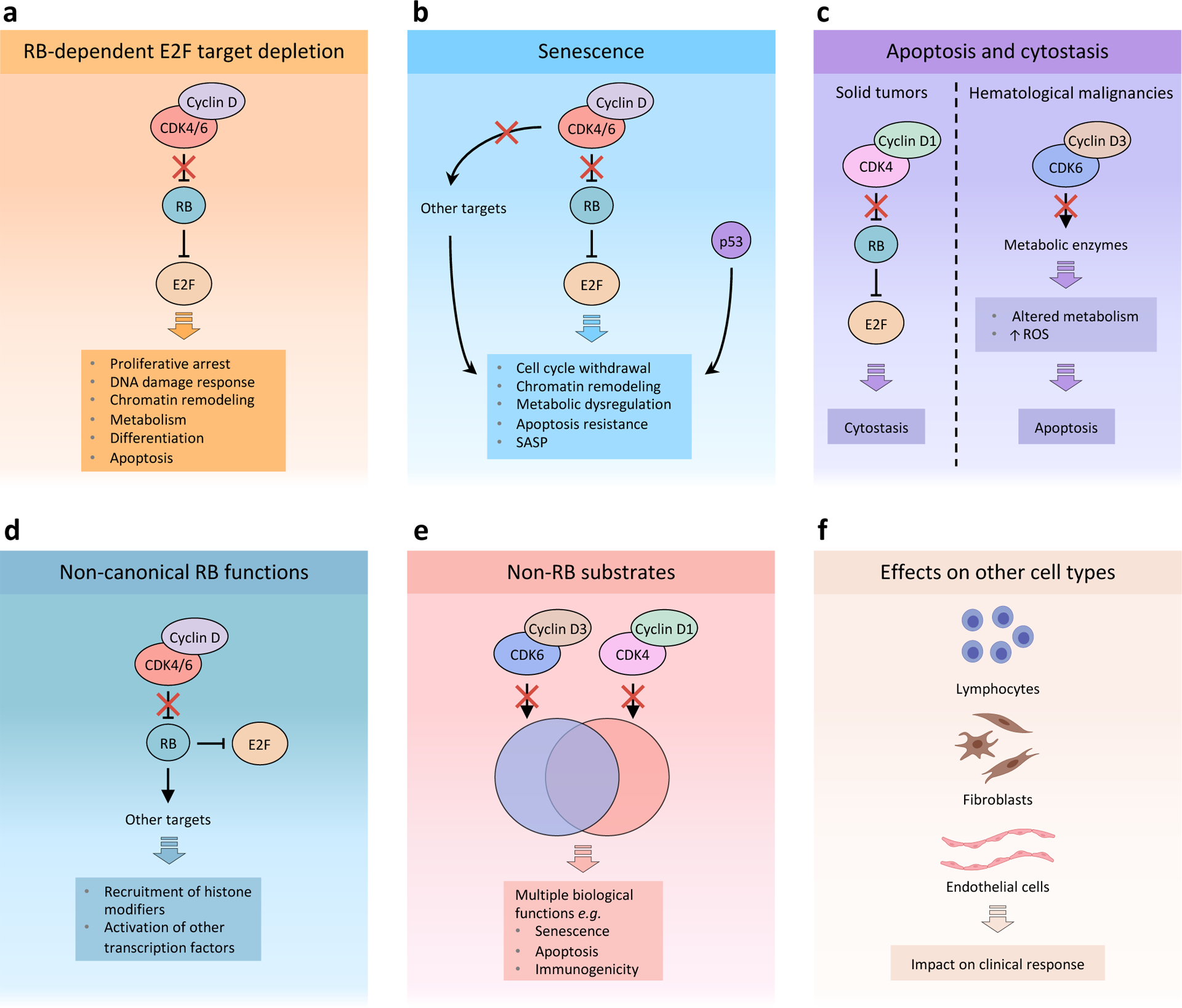
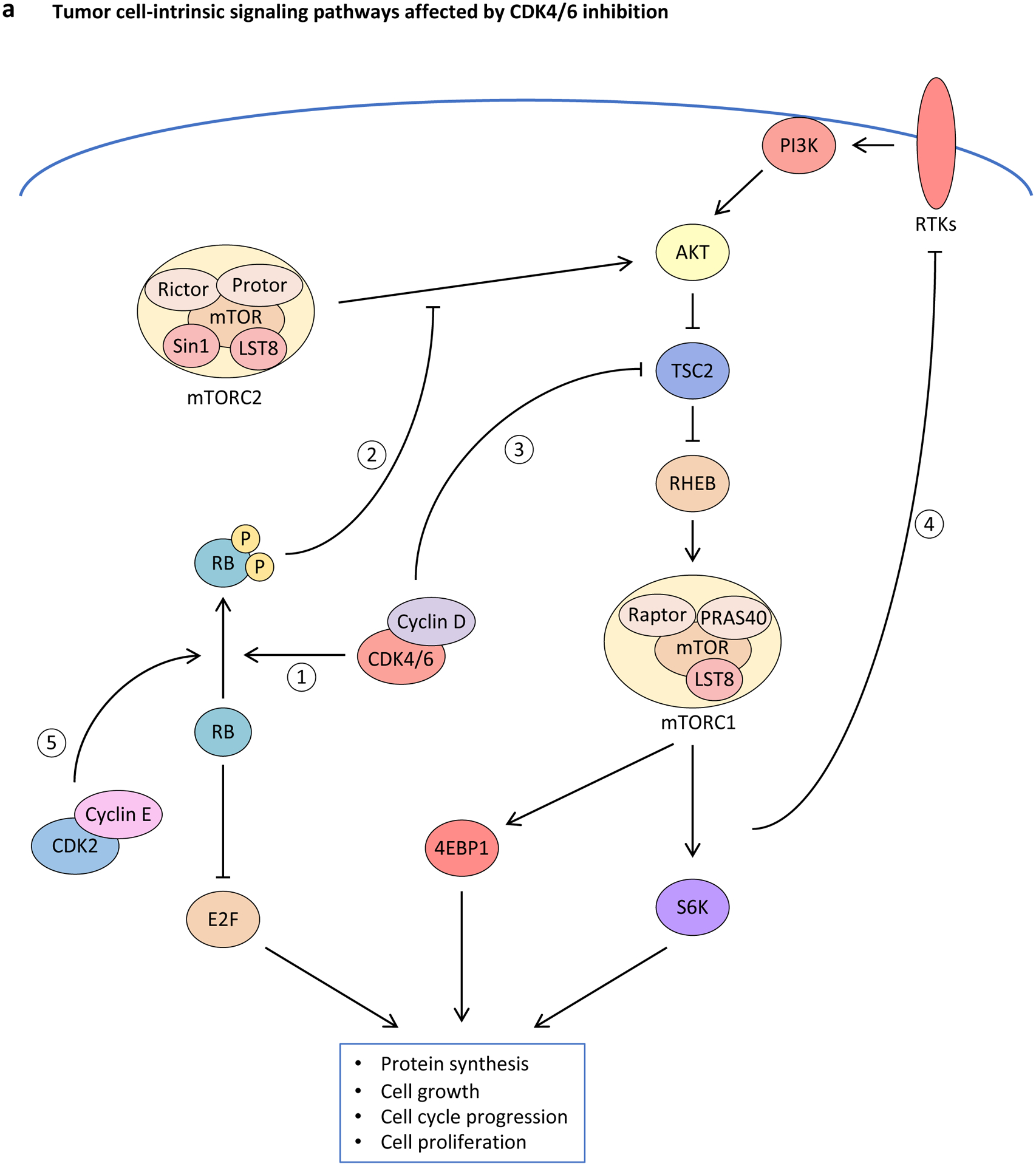
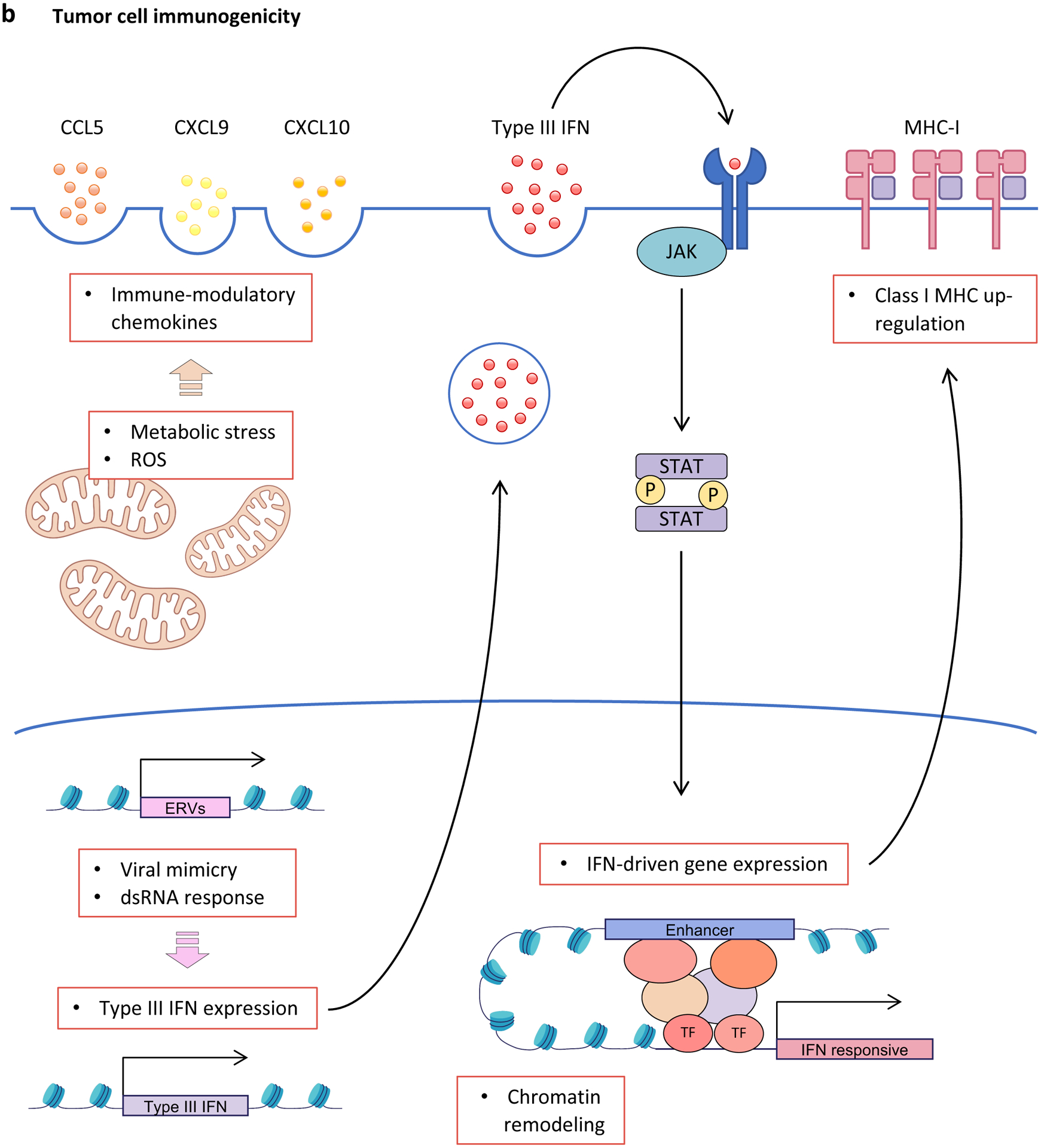
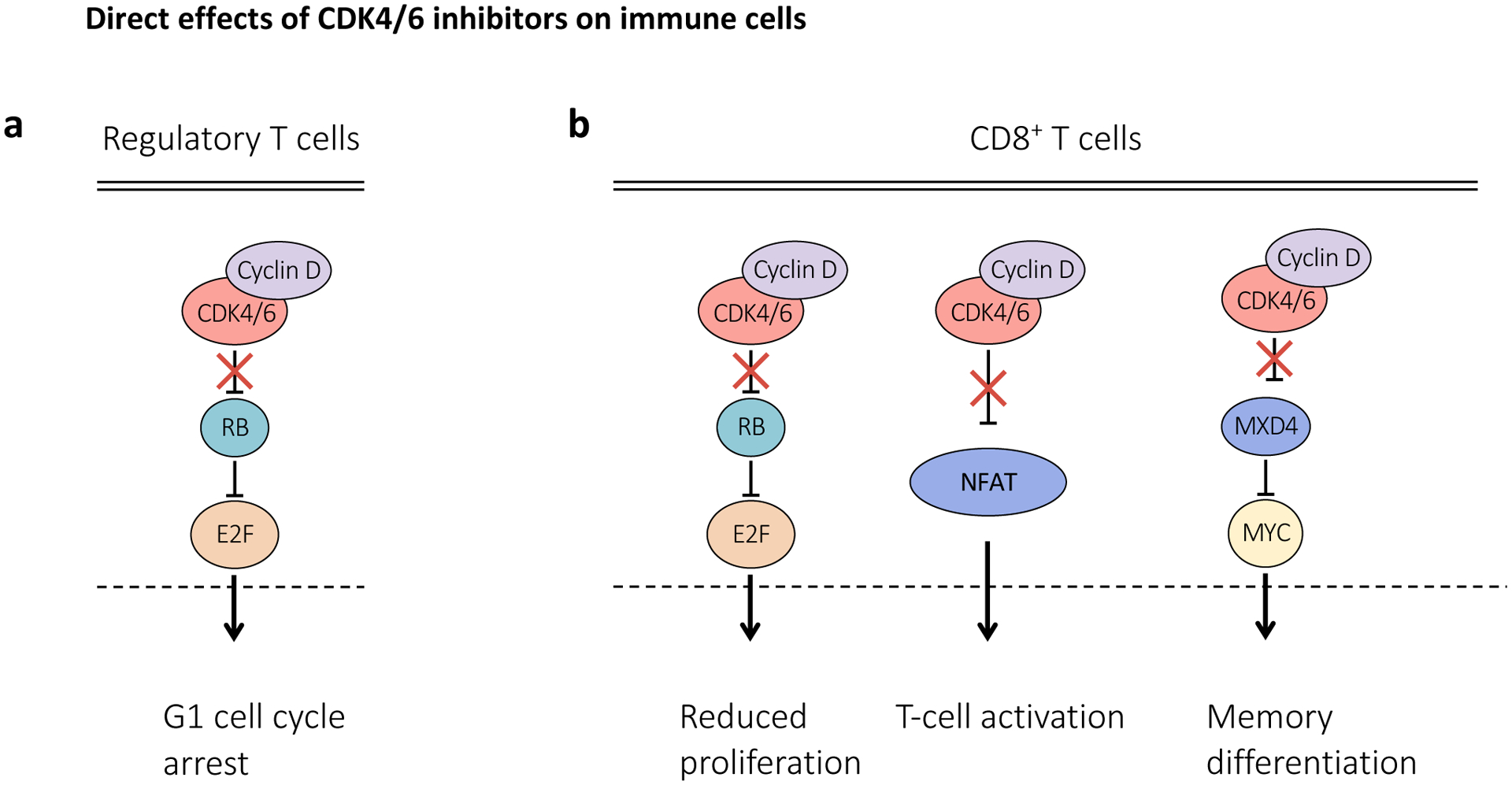
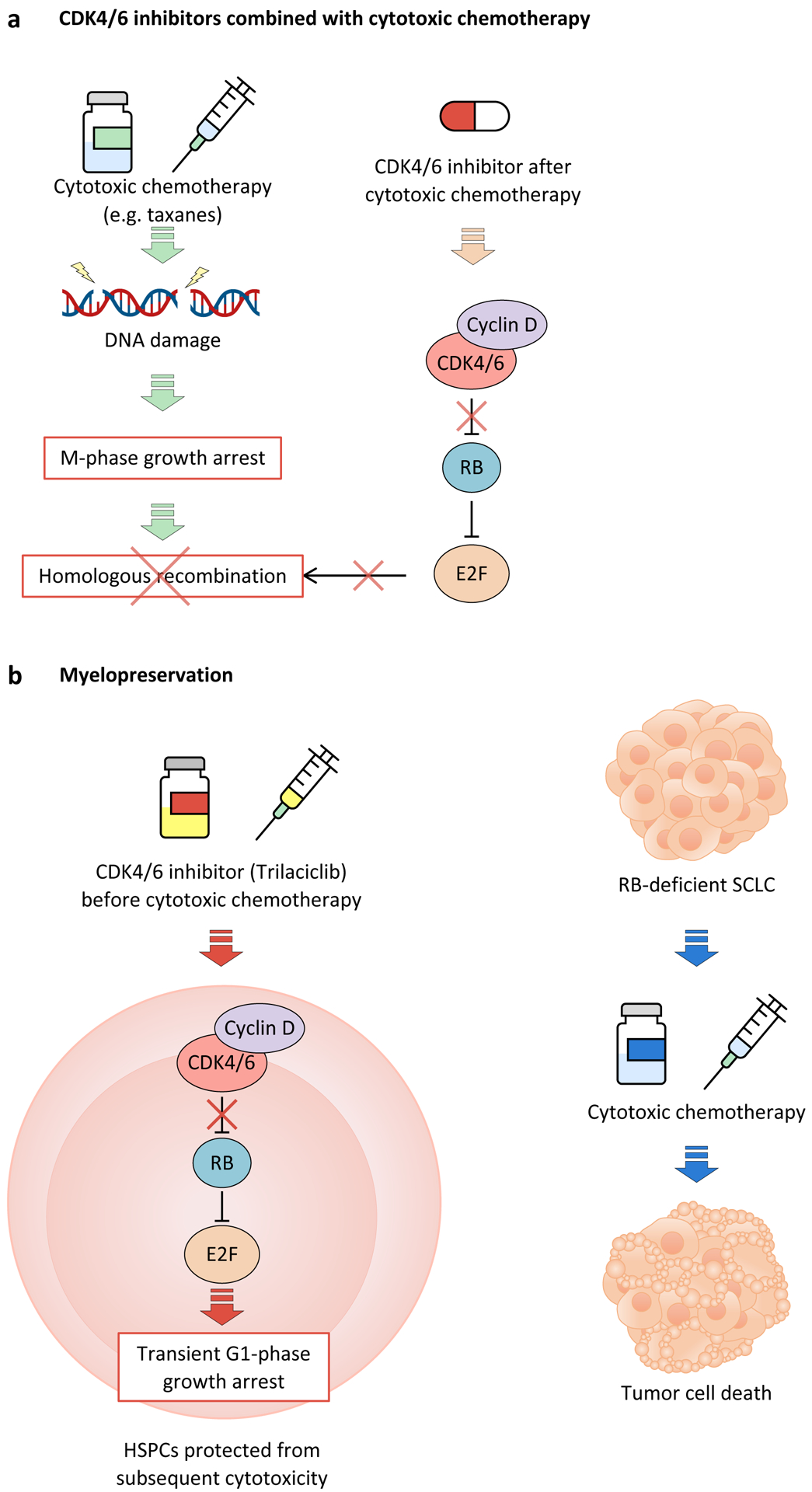
Cyclin-dependent kinase 4 (CDK4) and CDK6 are critical mediators of cellular transition into S phase and are important for the initiation, growth and survival of many cancer types. Pharmacological inhibitors of CDK4/6 have rapidly become a new standard of care for patients with advanced hormone receptor-positive breast cancer. As expected, CDK4/6 inhibitors arrest sensitive tumour cells in the G1 phase of the cell cycle. However, the effects of CDK4/6 inhibition are far more wide-reaching. New insights into their mechanisms of action have triggered identification of new therapeutic opportunities, including the development of novel combination regimens, expanded application to a broader range of cancers and use as supportive care to ameliorate the toxic effects of other therapies. Exploring these new opportunities in the clinic is an urgent priority, which in many cases has not been adequately addressed. Here, we provide a framework for conceptualizing the activity of CDK4/6 inhibitors in cancer and explain how this framework might shape the future clinical development of these agents. We also discuss the biological underpinnings of CDK4/6 inhibitor resistance, an increasingly common challenge in clinical oncology.
© 2022. Springer Nature Limited.
Figures
References
-
- Malumbres M et al. Mammalian Cells Cycle without the D-Type Cyclin-Dependent Kinases Cdk4 and Cdk6. Cell 118, 493–504 (2004). - PubMed
-
Using CDK4/CDK6 double knockout mice, this study established that mammalian cells can proliferate in the absence of both CDK4 and CDK6, demonstrating a plasticity of cyclin-CDK networks that has implications for CDK4/6 inhibitor resistance.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
