

Affinity2Vec: drug-target binding affinity prediction through representation learning, graph mining, and machine learning
- PMID: 35306525
- PMCID: PMC8934358
- DOI: 10.1038/s41598-022-08787-9
Affinity2Vec: drug-target binding affinity prediction through representation learning, graph mining, and machine learning
Abstract
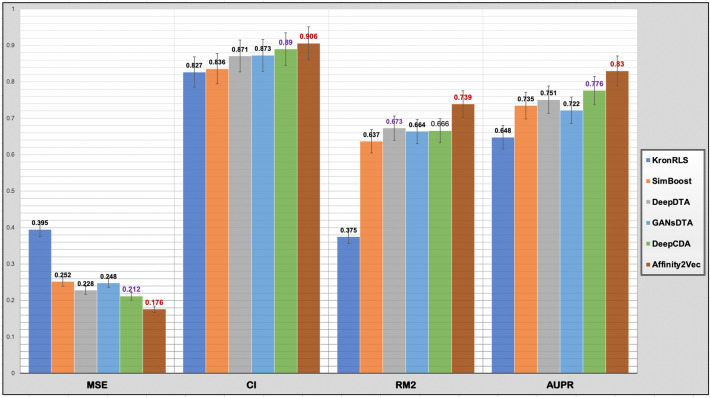
Drug-target interaction (DTI) prediction plays a crucial role in drug repositioning and virtual drug screening. Most DTI prediction methods cast the problem as a binary classification task to predict if interactions exist or as a regression task to predict continuous values that indicate a drug's ability to bind to a specific target. The regression-based methods provide insight beyond the binary relationship. However, most of these methods require the three-dimensional (3D) structural information of targets which are still not generally available to the targets. Despite this bottleneck, only a few methods address the drug-target binding affinity (DTBA) problem from a non-structure-based approach to avoid the 3D structure limitations. Here we propose Affinity2Vec, as a novel regression-based method that formulates the entire task as a graph-based problem. To develop this method, we constructed a weighted heterogeneous graph that integrates data from several sources, including drug-drug similarity, target-target similarity, and drug-target binding affinities. Affinity2Vec further combines several computational techniques from feature representation learning, graph mining, and machine learning to generate or extract features, build the model, and predict the binding affinity between the drug and the target with no 3D structural data. We conducted extensive experiments to evaluate and demonstrate the robustness and efficiency of the proposed method on benchmark datasets used in state-of-the-art non-structured-based drug-target binding affinity studies. Affinity2Vec showed superior and competitive results compared to the state-of-the-art methods based on several evaluation metrics, including mean squared error, rm2, concordance index, and area under the precision-recall curve.
© 2022. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures




References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Miscellaneous
